
Журнал «Здоровье ребенка» 1 (28) 2011
Вернуться к номеру
Лицелопаточно-плечевая миодистрофия Ландузи — Дежерина в клинике нейропедиатрии
Авторы: Кириллова Л.Г., Шевченко А.А., Яковлева С.М., Лисица В.В., Михайлец Л.П. ГУ «Институт педиатрии, акушерства и гинекологии АМН Украины», г. Киев
Рубрики: Педиатрия/Неонатология
Версия для печати
В статье приведен обзор литературы о редко встречающейся у детей патологии — прогрессирующей лицелопаточно-плечевой мышечной дистрофии Ландузи — Дежерина. Дано описание клинического случая пациента с отягощенным семейным анамнезом (11 случаев заболевания в роду), находившегося на обследовании и лечении в отделении психоневрологии ГУ «ИПАГ АМН Украины».
Мышечная дистрофия, атрофия, дети.
Лицелопаточно-плечевая миодистрофия Ландузи — Дежерина была описана Ландузи и Дежериным в 1884 г. Большинство отечественных детских неврологов впервые познакомились с данной патологией в руководстве Л.О. Бадаляна [1], которое до настоящего времени многим специалистам служит настольной книгой. При этом следует отметить и значительный вклад коллектива ученых, которые многие годы занимались и занимаются изучением диагностики, механизмов развития, клинических проявлений и лечения нейромышечных заболеваний [2, 5].
Миодистрофия Ландузи — Дежерина наследуется по аутосомно-доминантному типу с высокой пенетрантностью и встречается с частотой 0,9–2 на 100 000 населения. Установлена генетическая гетерогенность лицелопаточно-плечевой миодистрофии: в 90–95 % семей обнаруживается сцепление с локусом 4q35 (1A тип болезни), а в остальных 5–10 % — с локусом 10q26 (IB тип). Первичные биохимические дефекты в настоящее время неизвестны [4, 8, 11].
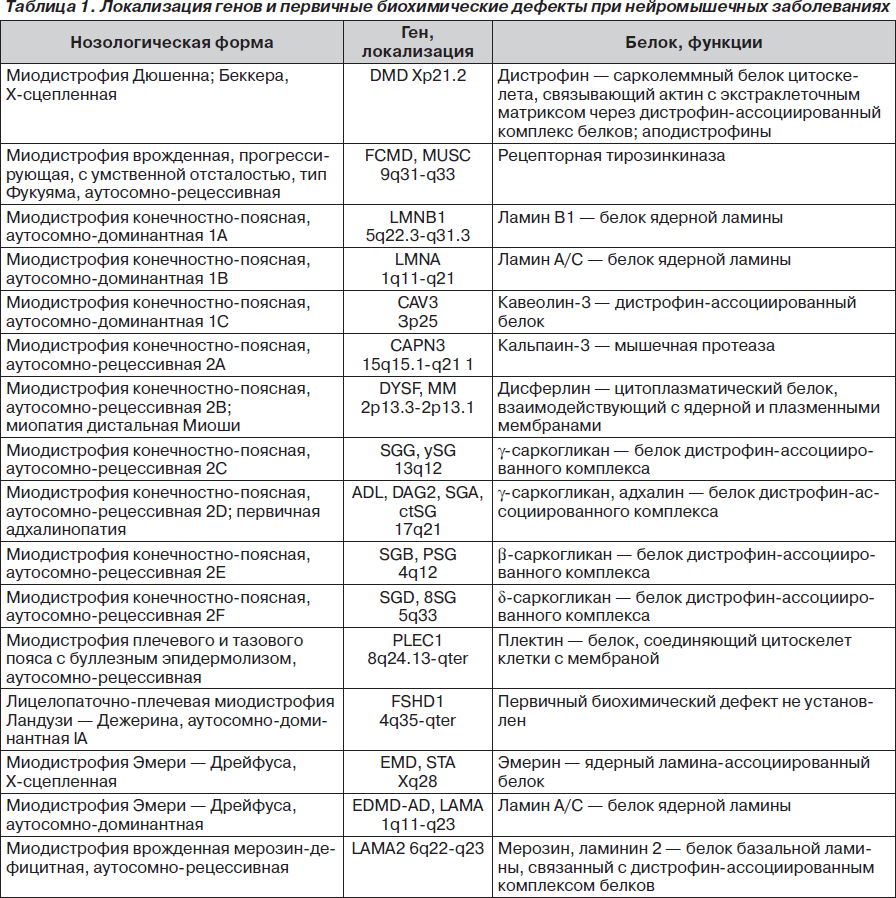
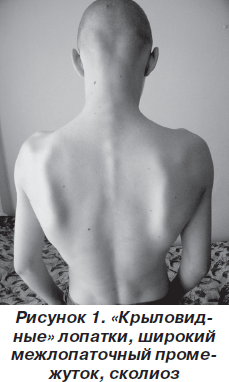
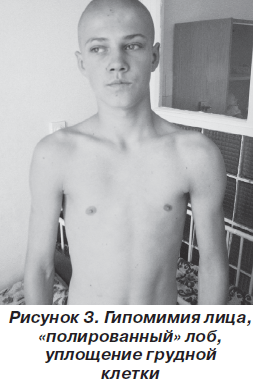
В классическом варианте первые признаки заболевания появляются в основном в возрасте 10–20 лет. Атрофии и мышечная слабость локализуются в области мимической мускулатуры, лопаток и плеч. Сначала атрофии наблюдаются в плечевом поясе, с последующим распространением на лицо. Обычно начальными проявлениями становятся затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании процесса грубо страдают круговые мышцы рта и глаз — не удается крепко зажмурить глаза и сжать губы. Вследствие атрофии лицо становится гипомимичным. Как правило, больные сами отмечают изменение своей мимики, их речь становится неразборчивой. Следует отметить и характерные симптомы в виде поперечной улыбки («улыбка Джоконды»), вывороченных губ («губы тапира»), «полированного» лба. Атрофии двуглавой и трехглавой мышц плеча, большой грудной, передней зубчатой, трапециевидной мышц могут обусловливать возникновение симптомов свободных надплечий, «крыловидных» лопаток, появление широкого межлопаточного промежутка, уплощения грудной клетки и сколиоза. В ряде случаев атрофии распространяются на мышцы ног. При этом слабость наиболее заметна в группе малоберцовых мышц по свисающей стопе, но может быть и в проксимальных отделах ног [5, 6, 10]. В зависимости от последовательности и характера распространения процесса выделяют собственно лицелопаточно-плечевую форму, лицелопаточно-плече-перонеальную, лицелопаточно-плече-ягодично-бедренную, лицелопаточно-плече-ягодично-бедренно-перонеальную и лицелопаточно-плече-перонеально-ягодично-бедренную формы. На ранних стадиях болезни мышечный тонус снижен в проксимальных группах мышц, глубокие рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча [1].
Необходимо отметить асимметричность атрофии, что является характерной клинической особенностьюданной патологии. В некоторых случаях имеет место псевдогипертрофия мышц. Контрактуры и ретракции выражены умеренно. Кардиомиопатия бывает в редких случаях. При проведении ангиоретинографии могут выявляться аномалии сосудов сетчатки. Во многих случаях при тяжелых глазных проявлениях находят телеангиэктазии, отек и отслойку сетчатки. Коагуляция телеангиэктазий предотвращает развитие слепоты. Может наблюдаться также и снижение слуха. Вышеперечисленные симптомы рассматриваются в качестве составляющей части фенотипических проявлений данной патологии. Тип течения болезни в большинстве случаев относительно благоприятный. Особо мы хотим акцентировать внимание на том, что физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни [9].
Несмотря на определенные успехи молекулярной генетики и другие достижения медицинской науки, диагностика данного заболевания на современном этапе основывается прежде всего на особенностях клиники (преимущественно лицелопаточно-плечевая локализация миодистрофического процесса) и на генеалогическом анализе (аутосомно-доминантный тип наследования).
Дифференциальную диагностику следует проводить с другими прогрессирующими мышечными дистрофиями (Эрба — Рота, Беккера и т.д.) и с вторичными миопатическими синдромами, которые возникают на фоне воспалительных, сосудистых, токсических и метаболических процессов.
Для уточнения диагноза используют биохимическое исследование с определением креатинфосфокиназы (КФК), электронейромиографию (ЭНМГ) и биопсию мышц. Уровень КФК может повышаться в 5 раз, но в некоторых случаях содержание фермента нормальное. На ЭНМГ регистрируются как миопатические, так и денервационные потенциалы. При проведении гистологических исследований во многих мышцах конечностей выявляют минимальные изменения, наибольшее число патологических признаков отмечается в надлопаточных мышцах, где обнаруживаются явления прогрессирующей дегенерации и небольшой краевой денервации [5, 7, 9].
Хотим обратить внимание на то, что в монографиях зарубежных авторов последних лет описана и инфантильная форма лицелопаточно-плечевой миодистрофии, которая быстро прогрессирует и приводит к тяжелой инвалидизации. Симптомы возникают еще в грудном возрасте, но не позднее 5 лет жизни в виде двустороннего пареза мимических мышц, что может имитировать врожденное поражение лицевых нервов. В дальнейшем развиваются ринолалия, а иногда птоз. Прогрессирующая проксимальная мышечная слабость возникает спустя 1–2 года после дебюта и в первую очередь захватывает плечи, а затем мышцы таза. Может отмечаться и псевдогипертрофия голеней. Сухожильные рефлексы снижены, а со временем исчезают. Отмечается быстрое нарастание слабости, что приводит к летальному исходу вследствие дыхательной недостаточности до достижения пациентом 20-летнего возраста. Очень редко в течение длительного срока слабость не прогрессирует, а тяжелая инвалидизация не наступает вплоть до зрелого возраста. У половины родственников больных обнаруживаются телеангиэктазии сетчатки и высокая частота снижения слуха. Диагноз данной патологии должен быть заподозрен у каждого ребенка с прогрессирующей лицевой диплегией, но в каждом случае необходимо исключить миастению и глиому мозгового ствола [8, 10].
Приводим таблицу, в которой представлены данные по локализации генов, ответственных за наследственные миопатии, с указанием первичных биохимических дефектов и краткой функциональной характеристикой этих белков (табл. 1) [12]. Как было отмечено выше, первичные биохимические дефекты в случае лицелопаточно-плечевой миодистрофии Ландузи — Дежерина в настоящее время неизвестны.
В настоящее время терапевтические возможности при миодистрофии Ландузи — Дежерина весьма ограниченны. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и на снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Терапевтический комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию, сбалансированное питание. В медикаментозном лечении прогрессирующих мышечных дистрофий следует отметить назначение курсами препаратов метаболического действия, направленных на улучшение и поддержание обменных, энергетических процессов неповрежденных миоцитов, кардиомиоцитов (кардонат, элькар, витамин Е, метионин, цитофлавин, АТФ-лонг, милдронат, кокарбоксилаза). Определенное значение имеет коррекция питания пациента. Рекомендуется диета с высоким содержанием белка и низким содержанием жиров, с пониженной калорийностью при оптимальном содержании витаминов и микроэлементов [5, 6, 8]. Важную роль играет и психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
В клинике детской психоневрологии ГУ «ИПАГ АМН Украины», где наряду с распространенными тяжелыми и труднодиагностируемыми неврологическими заболеваниями у детей также концентрируются и редкие неврологические нозологии, в течение последних пяти лет наблюдалось 7 пациентов с прогрессирующей миодистрофией Ландузи — Дежерина. Приводим описание истории болезни пациента, проходившего обследование и лечение в нашем отделении. Считаем, что этот клинический случай представляет значительный интерес, учитывая отягощенный семейный анамнез по данному заболеванию (11 случаев заболевания в роду).
Больной М., 16 лет, поступил в отделение с жалобами на слабость в верхних конечностях — невозможность поднять руки вверх. Слабость в руках больной отмечает на протяжении года, в то время как мать, страдающая аналогичным заболеванием, заметила снижение силы в верхних конечностях сына года три назад. Слабость постепенно нарастает. Заметное ухудшение отмечали после физических нагрузок — спортивных занятий на турнике и подъема тяжестей.
Из анамнеза жизни: мальчик от 2-й нормально протекавшей беременности, 2-х родов со слабостью родовой деятельности, родился с массой 4000 г, длиной 58 см. Развивался соответственно возрасту, учится в 9-м классе, удовлетворительно.
Неврологический статус: интеллект сохранен. Функция глазодвигательных нервов не нарушена. Выражена гипомимия лица и гипотрофия мышц плечевого пояса, «крыловидные» лопатки (рис. 1–3). Мышечная сила в проксимальных отделах верхних конечностей снижена до 3 баллов, больше слева, в дистальных — 5 баллов, в нижних конечностях — 3 балла. Мышечный тонус понижен. Сухожильные рефлексы на верхних и нижних конечностях живые, равномерные; брюшные рефлексы живые. Патологических стопных знаков нет. Походка не изменена. Координаторные пробы выполняет четко.

Обследование: общеклинические анализы без патологических изменений; активность ферментов сыворотки крови — АЛТ — 23 ед/л (N — 8–40 ед/л), АСТ — 34 ед/л (N — 10–41 ед/л), КФК — 148 (N < 270 ед/л); ЭНМГ — признаки первично-мышечного поражения.
Консультирован генетиком: подтвержден диагноз прогрессирующей мышечной дистрофии Ландузи — Дежерина.
Семейный анамнез: у матери пробанда, 41 года, в 14 лет началось заболевание, проявившееся слабостью мышц плечевого пояса; слабость мимической мускулатуры появилась с 35–36 лет. Аналогичное заболевание отмечается у ее отца, у 5 из 10 родных братьев и сестер и у 4 племянников (рис. 4). По месту жительства женщина получает социальное пособие в связи с прогрессирующей мышечной дистрофией Эрба — Рота.
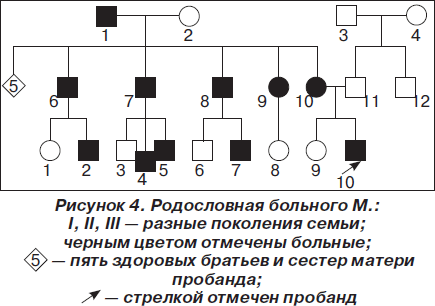
Анализируя семейный анамнез, обращаем внимание на то, что заболевание имеет наследственный характер с аутосомно-доминантным типом наследования. Ювенильная конечностно-поясная миодистрофия Эрба — Рота наследуется по аутосомно-рецессивному типу. Отличием клиники миодистрофии Эрба — Рота является преимущественно восходящий (начинающийся с нижних конечностей), а не нисходящий характер поражения, как наблюдается у всех больных членов описанной семьи. Поэтому считаем, что у мамы мальчика правомочен диагноз не миодистрофии Эрба — Рота, а миодистрофии Ландузи — Дежерина.
Представленный нами случай демонстрирует редкую форму прогрессирующей мышечной дистрофии с аутосомно-доминантным типом наследования — лицелопаточно-плечевую миодистрофию Ландузи — Дежерина. Особенностью случая является отягощенный семейный анамнез по данному заболеванию (11 больных в роду), что, однако, не вызвало настороженности медицинских работников при появлении первых симптомов у пациента. Важно отметить, что в случае своевременной диагностики и необходимого ограничения физических нагрузок возможно было бы избежать быстрого прогрессирования заболевания.
Таким образом, мышечные дистрофии характеризуются прогрессирующей мышечной слабостью, атрофией мышц и двигательными нарушениями. Однако различные формы миопатий отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками, что необходимо уметь своевременно диагностировать и дифференцировать. Очевидно, что в дальнейшем успех в лечении прогрессирующих мышечных дистрофий будет зависеть от новейших разработок в области молекулярной генетики, генной терапии. Поэтому вовремя назначенное лечение позволит предотвратить быстрое прогрессирование заболевания, улучшить качество жизни пациента и увеличить продолжительность его жизни.
В заключение хотим отметить, что публикация в широко читаемом педиатрическом журнале статьи, посвященной сугубо неврологической патологии, обусловлена тем, что больные первично обращаются не к неврологу, а к детским врачам и именно от их тактики во многих случаях зависит дальнейшая судьба пациентов.
Считаем необходимым напомнить, что еще в 60-е годы прошлого столетия сложными вопросами различных заболеваний нервной системы, в происхождении которых важную роль играет наследственная предрасположенность, занимался известный ученый с мировым именем С.Н. Давиденков, описавший лопаточно-перонеальную форму миопатий. В своих работах он впервые в Советском Союзе поднял проблему возможных прогнозов потомства при некоторых наследственных заболеваниях. С тех пор диагностика многих наследственных болезней поднялась на должную высоту благодаря новейшим молекулярно-генетическим технологиям. Однако способы определения типа наследования патологии остаются прежними, и каждому врачу важно помнить о том, что в случае доминантного типа «…риск передачи болезни потомству настолько велик, что больные члены семьи должны воздерживаться от деторождения. Наоборот, члены семьи, оставшиеся здоровыми, могут свободно иметь детей без всякой опасности передать им семейное заболевание» [4].
1. Бадалян Л.А. Детская неврология. — М.: Медицина, 1975. — 416 с.
2. Гехт Б.М. Нервно-мышечные болезни. — М.: Медицина, 1982. — 352 с.
3. Горбунова В.Н., Савельева Е.А., Красильников В.В. Молекулярная неврология. — СПб.: Интермедика, 2000. — Ч. 1. — 318 с.
4. Давиденков С.Н. Клинические лекции по нервным болезням. — Л.: Медгиз, 1961. — 360 с.
5. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко Л.Ф., Дегонская Е.В., Евтушенко И.С., Сохань Д.А. Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий // Международный неврологический журнал. — 2007. — № 4 (14). — С. 14-30.
6. Казаков В.М. Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями) // Неврол. журнал. — 2001. — № 3. — С. 47-52.
7. Armstrong D., Halliday W., Hawkins C. Pediatric neuropathology. A text-atlas. — Springer, 2007. — 426 p.
8. David R.B., Bodeinsteiner J.B., Mandelbaum D.E. Clinical pediatric neurology. — Demos Medical Publishing, New York, 2009. — 622 p.
9. Fenichel G.M. Clinical pediatric neurology. A signs and symptoms approach. — Saunders Elsevier, Philadelphia, 2009. — 415 p.
10. Menkes J.H., Sarnat H.B., Maria B.L. Child Neurology. — 7th Edition, Copyright-2006. — Lippincott Williams & Wilkins, 2006. — 1156 p.
11. Neuromuscular disorders: gene location // Neuromusc. Disord. — 1999. — Vol. 9, № 5. — P. 1-8.
12. http://www.bolezni-mishc.com/?p=1.