
Международный неврологический журнал 5 (43) 2011
Вернуться к номеру
Клінічний випадок міотонії Томсена у дитини
Авторы: Горб А.Л., КЗ «Херсонська обласна дитяча клінічна лікарня»
Рубрики: Неврология
Версия для печати
Хвороба Томсена відома клініцистам понад 130 років. Особливості клінічних проявів дозволяють своєчасно діагностувати захворювання. Поліморфізм міотонічних спазмів, особливо у період дебюту, може викликати труднощі у своєчасній верифікації діагнозу. У публікації наведено клінічний випадок міотонії Томсена з ізольованим міотонічним феноменом м’язів очей.
Міотонія, міотонічний феномен, м’язи, очі.
 Myotonia congenita, уроджена міотонія, хвороба Томсена — спадкове захворювання із дебютом у ранньому віці, що зберігається у хворого все життя.
Myotonia congenita, уроджена міотонія, хвороба Томсена — спадкове захворювання із дебютом у ранньому віці, що зберігається у хворого все життя.
Уперше опис захворювання був зроблений Лейденом у 1874 році. Томсен, який сам хворів на міотонію, у 1876 році вперше обґрунтував спадковий характер захворювання (на прикладі своєї сім’ї). Він спостерігав 20 хворих у чотирьох поколіннях, що також страждали на міотонічні розлади.
Міотонія (від грецького mio — м’язи, tonia — тонус) — нервово-м’язове захворювання, клінічними ознаками якого є м’язова гіпертрофія з наявністю міотонічного феномену (повільна релаксація м’язів після їх скорочення).
Тонічні спазми виникають після кожного інтенсивного м’язового скорочення та особливо виражені після періоду спокою, наприклад: спроба зімкнути повіки призводить до міотонічного скорочення кругових м’язів очей, що зберігається декілька секунд. Такий же феномен виникає при спробі зробити перші кроки після сидіння або при спробі розімкнути стиснуті пальці руки. Повторне скорочення одних і тих же м’язів супроводжується менш вираженим м’язовим спазмом, але у деяких випадках вираженість міо-тонічного феномену посилюється з кожним наступним м’язовим скороченням (парадоксальна міотонія).
Дебют захворювання може належати як до раннього віку (1–3 роки), так і дошкільного віку, з піком маніфестації у 8–10 років.
Першими симптомами можуть бути хибний симптом Грефе, що виникає через підвищення тонусу та порушення синкінезії повік та очних яблук, міотонічні реакції можуть відмічатися на акомодацію, при ковтанні, жуванні.
Згідно з класифікацією Беккера, виділяють п’ять типів домінантних міотоній (Becker, 1977):
І тип — класична міотонія Томсена;
ІІ тип — міотонія з флюктуючим перебігом;
ІІІ тип — типовою є холодова чутливість м’язів, особливо кругових м’язів очей та рота;
ІV тип — проміжний перебіг з переважним ураженням мімічних та окорухових м’язів;
V тип — ізольована перкусійна міотонія язика.
При об’єктивному обстеженні зазвичай патологічних змін черепно-мозкових нервів, чутливості, координації рухів не виявляється. У деяких випадках можливе зниження рефлексів та наявність гіпертрофій.
Діагностично важливими можуть бути симптоми «великого пальця» та «язика» — нанесення удару по тенору призводить до приведення І пальця, механічне подразнення язика перкусійним молотком призводить до виникнення ямки на язиці.
Прогресування повільне, у молодому віці може наставати стабілізація процесу. Міотонічні реакції протягом усього періоду захворювання у більшості випадків залишаються локальними, насамперед у мімічних, жувальних, ковтальних м’язах та у м’язах верхніх кінцівок.
Мікроскопічне дослідження біоптатів м’язів дозволяє виявити гіпертрофію м’язових волокон, наповнених міофібрилами.
Електронна мікроскопія — помірна гіпертрофія саркоплазматичного ретикулуму, зміна форми та розмірів мітохондрій, розширення телофрагми міофібрилярних волокон. Електроміографія дозволяє виявити повторні високочастотні розряди, при прослухуванні яких з’являється характерний звук «пікуючого бомбардувальника».
Типовими для міотонії Томсена є зменшення проникності мембран для іонів хлору та частково для іонів калію.
Міотонія Томсена спадкується за автосомно-домінантним типом. Спадковість обумовлена мутаціями в гені хлоридного каналу скелетних м’язів СLC № 1. Ген СLCN № 1 кодує один із хлоридних каналів м’язів СLC № 1 та розташований на довгому плечі сьомої хромосоми (7g 35). Ген, що кодує головний хлоридний канал, був клонований у 1991 році (Steinmeyer et al.). Поширеність — один випадок на 400 тис. сімей.
Вітальний прогноз задовільний. У більшості випадків хворі протягом життя соціально повністю адаптовані.
Специфічне ефективне лікування міотонії відсутнє. Ефективними можуть бути дієтичні заходи з обмеженням у харчовому раціоні продуктів із високим умістом калію (картопля, курага, ізюм, банани тощо).
Хворим рекомендуються дозовані фізичні навантаження, протипоказані купання у холодній воді, холодні напої та морозиво.
Клінічне спостереження. Дитина Ілля Г., 2004 року народження. Батьки дитини самостійно звернулися до інституту очних хвороб ім. Філатова у зв’язку з переміжною розбіжною косоокістю. Було рекомендовано неврологічне дообстеження. Дитина консультована у КЗ «Херсонська обласна дитяча лікарня». Скарги на епізодичну розбіжну косоокість, часте кліпання повіками при напруженні.
Дитина від першої вагітності, загроза викидню у ІІ триместрі. Пологи у 40 тижнів, фізіологічні, вага при народженні — 3500 г, оцінка за шкалою Апгар — 9 балів. Статокінетичний, мовленєвий, психічний розвиток за віком.
У 2,5 року батьки звернули увагу на переміжну розбіжну косоокість, яка мала тенденцію до прогресування, переважно лівого ока.
При неврологічному обстеженні — когнітивні функції за віком, зіниці D = S, окорухових порушень не встановлено, слабкість конвергенції ОS. Функції мімічних м’язів збережені, бульбарні та псевдобульбарні порушення відсутні. М’язовий тонус збережений, без патологічних змін. Рефлекторних порушень не встановлено. Патологічні симптоми відсутні.
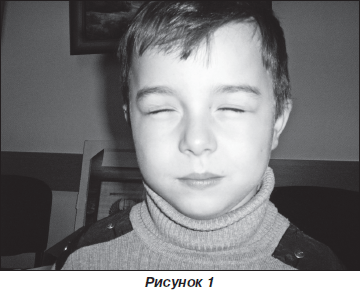
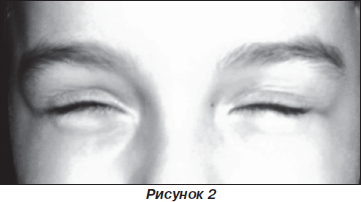
При проведенні проби Ромберга діагностовано міо-тонічний феномен (після виконання проби у дитини виникли труднощі розплющити очі) (рис. 1, 2).
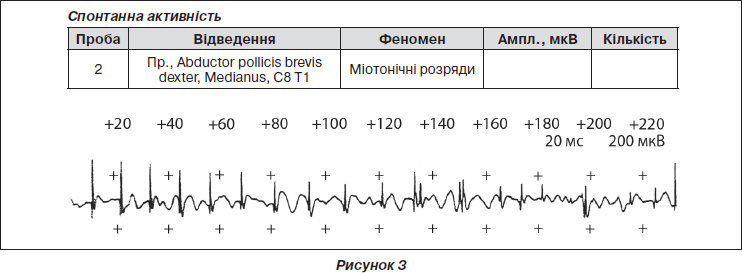
Дитині призначено ЕМГ. Висновок — m.Abductor pollicis brevis dexter реєструються множинні міотонічні розряди (як спонтанно, так і при переміщенні електрода). ПДЄ змінені за гіпертрофічним типом (рис. 3).
Офтальмоскопія: очне дно без патологічних змін. Збіжна косоокість співдружня, неакомодаційна, альтернуюча.
Клініко-біохімічні аналізи крові, сечі без патологічних змін.
Рівень креатинфосфокінази, лактатдегідрогенази не перевищує фізіологічну норму.
ЕКГ: ритм синусовий, вертикальне положення електричної осі серця.
Допплер-ЕхоКГ: камери серця не розширені, товщина міокарда в нормі, перегородки відстежені по всій довжині, функція клапанів у нормі.
Дитині встановлено діагноз: міотонія Томсена.
Спадковість — батько хлопчика хворіє на міотонію Томсена з класичним перебігом.
Перші ознаки захворювання з’явились у 5-річному віці, виникли тонічні спазми м’язів нижніх кінцівок при фізичних навантаженнях. У подальшому тонічні спазми з’явилися у верхніх кінцівках. У підлітковому віці відмічались виражені гіпертрофії м’язів шийно-плечової зони, які згодом зменшились.
У теперішній час міотонічні феномени зберігаються тільки у верхніх кінцівках. Соціально адаптований.
Висновки
Своєчасна діагностика вродженої міотонії — це, насамперед, задовільний прогноз щодо розвитку когнітивних та рухових функцій з відсутністю інвалідизуючих обмежень здоров’я у майбутньому, а в деяких випадках — це захист дитини від хибних діагнозів та необґрунтованого їх лікування.
1. Брильман Джон, Коэн Скотт. Неврология. — Москва, 2007. — С. 144-146.
2. Вельтищев П.А., Темин П.А. Наследственное заболевание нервной системы. — Москва: Медицина, 1998.
3. Иллариошкин С.М., Иванова-Смоленская И.Л., Маркова Е.Д. ДНК-диагностика и медико-генетическое консультирование в неврологии. — Москва: МИА, 2002.
4. Карлов В.А. Неврология. — Москва, 2002. — С. 528-531.
5. Сепп Є.Н., Цукер М.Б., Шмід Є.В. Нервові хвороби. — Київ: Держмедвидав, 1951. — С. 453-455.
6. Шмид Е.В. Учебник нервных болезней. — Москва, 1942. — С. 472-474.
7. Цукер М.Б. Клиническая неврология детского возраста. — Москва: Медицина, 1986.