
Международный неврологический журнал 6 (44) 2011
Вернуться к номеру
Роль генеалогических факторов в патогенезе болезни Паркинсона
Авторы: Карабань Н.В., ГУ «Институт геронтологии им. Д.Ф. Чеботарева» НАМН Украины, г. Киев
Рубрики: Неврология
Версия для печати
Обследовано 342 пациента с болезнью Паркинсона (БП) в возрасте 50–70 лет с длительностью заболевания от 3 до 15 лет и 74 клинически здоровых родственника в возрасте до 45 лет. При клинико-генеалогическом анализе 52 семей больных БП установлены аутосомно-доминантный (76,6 % случаев) и аутосомно-рецессивный (23,4 % случаев) типы наследования заболевания. Нейрофизиологическим критерием определения вероятности наследования БП от пробандов к родственникам 1-й степени родства является ЭМГ-диагностика, согласно которой происходит наследование экстрапирамидного типа нарушений центрального супраспинального контроля мышечного тонуса в 48 % случаев, что может рассматриваться как фактор риска развития БП. У клинически здоровых соблизнецов регистрируются ЭМГ-признаки экстрапирамидной недостаточности, совпадающей со сторонностью гемисиндрома паркинсонизма у больного близнеца.
Патогенез болезни Паркинсона (БП), как и других нейродегенеративных заболеваний, обусловлен комплексом патологических процессов, включающих митохондриальную дисфункцию, оксидативный и протеолитический стрессы, локальное воспаление, апоптоз. Эти процессы могут быть детерминированы различными генетическими, внешнесредовыми, а также возрастными инволютивными факторами и их взаимодействием [12, 26].
В настоящее время существует несколько точек зрения относительно генетической предрасположенности к возникновению болезни Паркинсона. Эпидемиологические данные показали вдвое большую вероятность получить это заболевание у родственников первой степени родства по сравнению с контролем. В проспективном исследовании 211 пациентов с БП отмечено семейное наследование заболевания в 22 % случаев. Если наличие семейных кластеров можно объяснить проживанием родственников в одной и той же среде, то у некоторых из них прослеживается аутосомно-доминантный тип наследования заболевания с мутацией в гене a-синуклеина, вызывающей БП [1].
Для определения генетического вклада в БП были широко использованы исследования близнецов [2, 6], при этом W.J. Johnson и соавт. [2] отметили противоречивость результатов исследований генетической предрасположенности к БП. Так, позитронно-эмиссионная томография (ПЭТ) с применением поглощения меченой 18F-флуородопы в путамене у 16 асимптомных близнецов с БП продемонстрировала уровни конкордантности дефицита дофамина в 45 % для монозиготных близнецов и в 29 % — для дизиготных близнецов, что значительно выше показателей, определяемых при клиническом исследовании (8 и 5 % соответственно). У всех асимптомных близнецов отмечено достоверное снижение поглощения меченой флуородопы в стриатуме при ПЭТ-исследовании. Эти противоречивые результаты показывают необходимость дополнительных клинических обследований близнецов для сравнительного определения относительного вклада генов и факторов среды в развитие спорадической БП [3].
Близнецовые и семейные исследования основывались на определении связи заболевания с наследственностью лишь на основе анамнеза и не дифференцировали БП от симптоматического паркинсонизма. ПЭТ-исследование поглощения 18F-флуородопы в семи генетически не связанных между собой семьях, в которых было двое больных БП, продемонстрировало снижение поглощения флуородопы в путамене у 8 из 32 (25 %) клинически бессимптомных членов семей [4]. Таким образом, снижение выраженности симптоматики при болезни с более поздним началом может маскировать существующую генетическую предрасположенность к БП.
При болезни Паркинсона положительный семейный анамнез выявляется у 10–15 % больных [5], однако абсолютное большинство случаев БП являются спорадическими. У этих больных заболевание имеет мультифакториальную природу и является результатом взаимодействия разнообразных генетических и средовых факторов. Имеющаяся генетическая предрасположенность к БП определяется совокупным действием ряда генов, контролирующих активность дофаминергической трансмиссии, процессы детоксикации и клеточной энергетики [6]. Вклад этих генов, детерминирующих определенный метаболический фон у близких родственников, а также сходный характер разнообразных экзогенных воздействий у членов одной семьи лежат в основе нередко наблюдающегося накопления повторных случаев БП в обследуемых семьях. Подтверждением этому являются результаты эпидемиологических исследований, показавших, что родственники больных с данным заболеванием имеют увеличенный, в среднем в 2–7 раз, риск развития БП по сравнению с контрольной группой [7], причем более высокий риск выявлен для родственников больных с ранним началом болезни.
В литературе описано небольшое число семей, в которых классическая БП с тельцами Леви, подтвержденная на секции, наблюдалась у ряда родственников из нескольких поколений и наследовалась как моногенный менделевский признак. В большинстве таких описанных родословных отмечен аутосомно-доминантный тип наследования [5, 6, 8]. Несмотря на большую редкость таких наследственных случаев БП, именно они благодаря современным ДНК-технологиям стали основным объектом исследования, позволившим идентифицировать ключевые молекулярные механизмы патогенеза БП, применяемые как для семейных, так и для спорадических случаев заболевания.
Генетические локусы семейных форм БП весьма многообразны, но на сегодняшний день наиболее изученными являются 6 генетических вариантов первичного паркинсонизма (табл. 1).
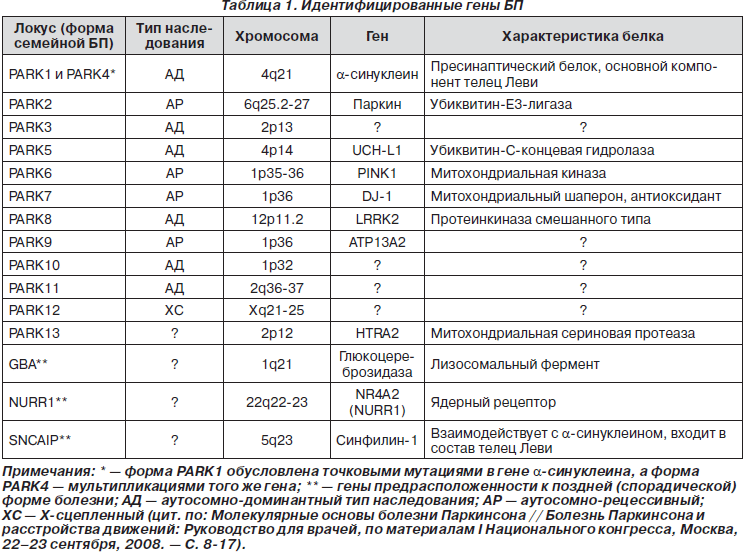
PARK1 (a-синуклеин). Альфа-синуклеин представляет собой высококонсервативный компактный мономерный белок, практически не имеющий вторичной структуры. Он является пресинаптическим белком, который связан с везикулярными и мембранными структурами и, предположительно, играет роль в синаптическом везикулярном транспорте, хранении и компартаментализации нейротрансмиттеров [10, 11]. В единичных семьях описаны 3 точковые миссенс-мутации в гене a-синуклеина, однако чаще при PARK1-форме БП (а также в части случаев болезни диффузных телец Леви) встречается полная мультипликация соответствующей области хромосомы 4q21 [9]. Белок a-синуклеин имеет повышенную склонность к агрегации, а в фибриллярном полимеризованном виде он является основным компонентом телец Леви [12, 13]. В настоящее время a-синуклеин рассматривается в качестве ключевого молекулярного маркера патологии нейронов и модулятора процессов нейродегенерации паркинсоновского типа.
PARK2 (паркин). Мутации в гене паркина являются наиболее частой причиной раннего (в том числе ювенильного) аутосомно-рецессивного паркинсонизма — более 50 % семейных и около 15 % спорадических случаев с ранним началом симптомов [10, 11]. Высказывается предположение, что носительство мутаций паркина даже в гетерозиготном состоянии может являться значимым (хотя и не абсолютным) фактором риска развития БП [11, 14]. Паркин представляет собой убиквитин-протеинлигазу типа Е3, функция которой заключается в доставке аномально конформированных белков в протеасомный комплекс для последующего расщепления [12]. Именно с утратой убиквитин-лигазной активности связывают развитие аутосомно-рецессивной PARK2-формы первичного паркинсонизма.
PARK6 (PINK1). Данная форма аутосомно-рецессивного паркинсонизма обусловливает от 1 до 9 % всех случаев паркинсонизма с ранним началом симптомов [15].
PARK7 (DJ-1). PARK7-форма аутосомно-рецессивного паркинсонизма является редкостью: около 1–2 % случаев ранней БП вызваны мутациями данного гена [16]. Белок DJ-1 эволюционно консервативен, локализован в митохондриях и обладает предположительно активностью антиоксиданта (работающего по принципу «сборщика» реактивных форм кислорода), транскрипционного коактиватора и митохондриального шаперона [17].
PARK8 (LRRK2). Данный ген связан с аутосомно-доминантной формой паркинсонизма, его пенетрантность составляет около 40 %. Ген LRRK2 (Leucine-rich repeat kinase 2) имеет чрезвычайно большое значение в развитии БП в общей популяции. Показано, что те или иные мутации в нем обусловливают 1–7 % всех спорадических и семейных случаев БП в европейских популяциях и 20–40 % — в некоторых популяциях ближневосточно-средиземноморского региона [18–20]. Спектр PARK8-ассоци- ированных фенотипов весьма широк — от типичной поздней БП с тельцами Леви до атипичных вариантов синуклеин- и таупатологии.
Ген GBA. Данный ген кодирует лизосомальный фермент глюкоцереброзидазу и традиционно связывается с развитием хорошо известной аутосомно-рецессивной формы лизосомной болезни накопления — болезни Гоше. Однако значительно позже было установлено, что в семьях, отягощенных болезнью Гоше, достоверно чаще по сравнению с общей популяцией обнаруживаются случаи БП [21]. Дальнейший анализ подтвердил связь гетерозиготного носительства мутаций в гене GBA с высоким риском развития различных вариантов синуклеинопатий — классической БП и деменции с тельцами Леви [11].
Если близнецовые исследования не ответили на вопросы об относительном влиянии генов и среды на развитие спорадической БП, то некоторое увеличение риска заболеваемости в семьях указывает на сложное полигенное наследование. Трудно быть уверенным в генетическом влиянии на развитие БП у больных с поздним началом, однако возможно, что несколько функциональных полиморфных генов, действующих совместно, создают предрасположенность к этому заболеванию. Роль средовых и генетических влияний в патогенезе БП заключается в том, что в результате первичного воздействия определенных средовых токсинов изменения нескольких полиморфных генов вторично повышают риск дегенерации ДА-нейронов [8, 22].
В молекулярных механизмах БП значительную роль играет нарушение системы контроля за биогенезом, пространственной организацией и биодеградацией нейрональных белков [12]. При этом ключевая роль отводится a-синуклеину, патологическая агрегация которого в виде фибриллярных структур является первым этапом формирования в дегенерирующих нейронах телец Леви.
Согласно современным представлениям, агрегированный a-синуклеин является прямым медиатором нейротоксичности в дофаминергических нейронах [23].
Эта нейротоксичность реализуется за счет различных механизмов: индукции свободнорадикальных реакций, блока везикулярного транспорта из эндоплазматического ретикулума в аппарат Гольджи, активации стрессовых протеинкиназ и процессов апоптоза, микроглии, нарушения взаимодействия аномального a-синуклеина с его естественными белковыми «партнерами» — синфилином-1, актином, тубулином, ГФДГ и др. [24].
Понимая всю сложность проблемы изучения роли наследственного фактора в генезе БП, мы ограничили свои исследования клинико-генеалогическим и ЭМГ-анализом типов наследования БП у больных среднего и пожилого возраста, находящихся на диспансерном наблюдении в Отделе клинической физиологии патологии экстрапирамидной нервной системы Института геронтологии НАМН Украины.
Для этого были поставлены следующие задачи: 1) уточнить наличие наследственного фактора в возникновении БП; 2) установить тип наследования в семейных случаях заболевания; 3) провести анализ внутрисемейных клинических особенностей БП; 4) показать диагностические возможности ЭМГ у лиц из группы риска.
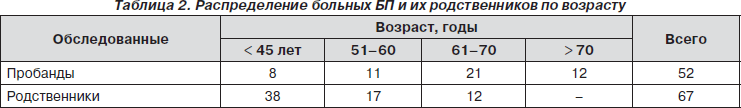
Обследуемые и методы. Проведено клинико-электромиографическое обследование 342 пациентов с БП в возрасте 50–70 лет с длительностью заболевания от 3 до 15 лет и 74 клинически здоровых родственников в возрасте от 23 до 45 лет, а также клинико-генеалогическое наследование БП в 52 семьях больных и у 67 их родственников.
Биоэлектрическая активность мышц регистрировалась на 4-канальном электромиографе Medicor MG 440 при полосе пропускания 10 Гц – 10 кГц. Через аналогово-цифровой преобразователь сигналы поступали в компьютер IBM PC и записывались на жесткий диск при частоте выборки по каждому каналу 1000 Гц и длительности каждой записи 10 с. Записывали поверхностную ЭМГ m. biceps brachii и m. triceps brachii с помощью биполярных электродов (0,5 1,0 см2) с постоянным межэлектродным расстоянием 1,5 с. Регистрировали состояние мышц в покое, при синергических изменениях мышечного тонуса, произвольных максимальных по силе сокращениях исследуемых мышц в пробах со сгибанием и разгибанием. Определялись тип ЭМГ, частота (Гц) и амплитуда (мкВ) колебаний. При оценке типологических особенностей общей структуры ЭМГ использовали классификацию Ю.С. Юсевич (1972). Повышение амплитуды токов в покое выше 10,5 мкВ и появление залповой активности на ЭМГ (III тип по Юсевич) относили к признакам экстрапирамидной недостаточности.
Результаты и обсуждение. Проведено клинико-генеалогическое обследование 52 семей, в которых обнаружена БП у пробандов, что позволило составить их общую характеристику (табл. 2). Установлено, что у пробандов равномерно встречаются случаи аутосомно-доминантного наследования и спорадических случаев возникновения заболевания.
У больных с аутосомно-доминантным типом наследования БП, как правило, отмечалось более молодое начало болезни, преобладание акинетико-ригидного синдрома, что считается клинически и прогностически более неблагоприятным. У родственников больных БП при обоих типах наследования клинически определялись отдельные признаки ЭПН и в нескольких случаях — болезнь Паркинсона.
ЭМГ-диагностика выявила преобладание І–ІІІ типа биоэлектрической активности мышц, который в нейрофизиологической литературе рассматривается как условно-экстрапирамидный [25, 26].
Большинство обследованных больных были в возрасте от 50 до 70 лет (табл. 2). Длительность заболевания у пробандов колебалась от 3 до 15 лет, в то время как преобладающее количество обследованных родственников (38 чел., 56,7 %) было в возрасте до 45 лет, т.е. моложе возраста типичного начала БП.
При аутосомно-доминантном типе наследования БП наследовалась пробандом-мужчиной от матери, а у пробанда-женщины — от отца.
Под нашим наблюдением находились 4 семьи, в которых муж и жена независимо друг от друга заболели БП в пожилом возрасте. При клинико-генеалогическом обследовании их детей в 42 % наблюдений отмечены отдельные симптомы экстрапирамидной недостаточности, такие как мелкоамплитудный гемитремор покоя и легкая ригидность, совпадающие по сторонности проявления с подобными симптомами у больных родителей-пробандов.
Особый интерес представляют генетически детерминированные случаи БП у близнецов. Впервые на эти признаки обратил внимание D. Brooks [3], который показал, что ПЭТ, проведенная у 16 асимптомных соблизнецов больных БП, выявила уменьшение поглощения меченой 18F-флуородопы в путамене на 45 % в группе монозиготных и на 29 % — в группе дизиготных близнецов. Эти данные с высокой степенью доказательности свидетельствуют о значении генетического фактора в генезе БП.
Мы имели уникальную возможность наблюдать клинические проявления БП у 2 пар монозиготных близнецов. В качестве иллюстрации приводим клинические описания этих случаев и записи ЭМГ у больных БП и практически здоровых соблизнецов.
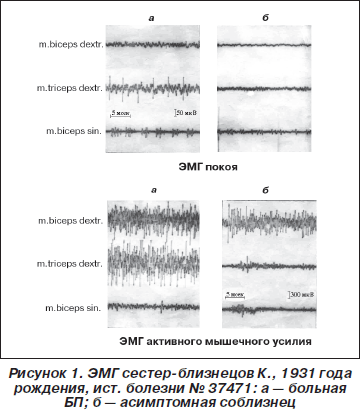
Сестры-близнецы К., 1931 г.р., история болезни № 37471. Анамнез заболевания у сестры Л.Д. в течение 9 лет. В неврологическом статусе преобладает дрожательно-акинетико-ригидный синдром 2,0 ст. (по Хен — Яру), правосторонний гемитип. Заболевание медленно прогрессировало, ввиду чего заместительную терапию леводопасодержащими препаратами больная стала принимать только в последние 2 года. Соблизнец Р.Д. практически здорова, работает врачом-педиатром. Обе сестры происходят из рода долгожителей: отец умер в 92 года, у него дрожали руки; мать умерла в 89 лет. У тети по отцу также дрожали руки. Проведенное ЭМГ-исследование показало (рис. 1) наличие залповой активности у здоровой сестры как в фоновой записи, так и при ЭМГ активного мышечного усилия.
Братья-близнецы Я., 1946 г.р., история болезни № 53419. Анамнез болезни у брата Д.А. в течение двух лет: заболевание началось со скованности в правой руке, которая значительно усилилась после перенесенного стресса. В неврологическом статусе доминировали акинетико-ригидный правосторонний гемитип 1,5 стадии (по Хен — Яру), начальные нарушения фонации, выраженная гипомимия. Соблизнец В.А. практически здоров, работает мастером на заводе. Дети обоих близнецов здоровы, от ЭМГ-обследования отказались по этическим причинам. У матери этих братьев-близнецов дрожали руки, умерла в 90 лет от сердечной недостаточности. На ЭМГ у здорового соблизнеца выраженная залповая активность, характерная для ІІІ типа, усиливающаяся при ближних и дальних синергиях (рис. 2).
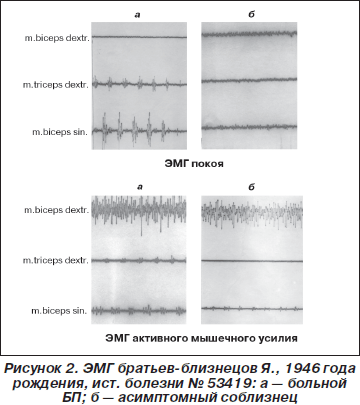
Следует полагать, что ЭМГ в этих случаях явилась высокоинформативным методом диагностики БП и латентной экстрапирамидной недостаточности (ЭПН).
ЭМГ в диагностике наследственной предрасположенности к БП. Известно, что по мере старения наблюдается увеличение амплитуды биопотенциалов покоя, что может отражать субклиническую стадию проявлений возрастного тремора рук и свидетельствовать об ослаблении тормозных надсегментарных влияний с возрастом [25, 26]. Показано, что регистрация залповой активности на ЭМГ у практически здоровых лиц может косвенно отражать формирование скрытого генератора патологически усиленного возбуждения в стриатуме [26].
Важным преимуществом ЭМГ-метода для диагностики БП считается его информативность для выявления субклинических и начальных проявлений заболевания. Установлено, что ЭМГ при БП характеризуется высоковольтными колебаниями биопотенциалов мышц в покое по типу залпов (залповая активность). Этот вид ЭМГ-активности опре-деляется как ІІІ тип по Ю.С. Юсевич. ЭМГ ІІІ типа регистрируется в 17,3 и 26,2 % случаев и у клинически здоровых людей среднего и пожилого возраста, что может отражать наличие скрытой ЭПН, которую можно рассматривать как фактор риска развития БП [26].
Установлено, что при регистрации поверхностной ЭМГ в состоянии покоя у всех клинически здоровых лиц независимо от возраста наблюдается І тип ЭМГ. К наиболее характерным изменениям ЭМГ при БП относят высокий уровень биоэлектрической активности мышц в покое и залповую активность, соответствующую ритму дрожания [25]. Длительность залпов обычно колеблется от 40–160 мс, а амплитуда их была в пределах 50–250 мкВ, что в 3–4 раза превышает норму.
Отдавая должное приведенным данным литературы о структуре и генезе ЭМГ-изменений при старении и паркинсонизме, мы поставили перед собой задачу показать возможность применения ЭМГ в качестве одного из диагностических критериев для определения возможности наследования экстрапирамидного типа нарушений центрального супраспинального контроля мышечного тонуса от пробанда к родственникам. Получена качественная характеристика типов ЭМГ (формирование которой отражает структурные основы биоэлектрической активности мышц) у 342 больных БП и 74 их клинически здоровых родственников (табл. 3). Обнаружено, что у 48 % родственников больных обнаружен ІІІ тип ЭМГ, что можно рассматривать как проявление генетически детерминированной ЭПН, являющейся фактором риска развития паркинсонизма.
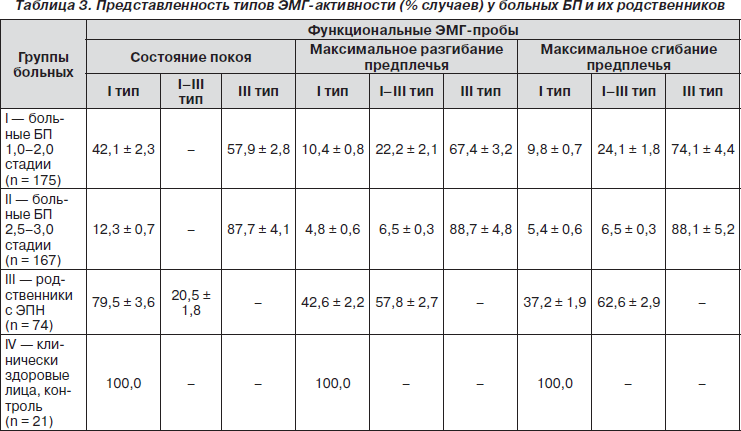
Из табл. 3 видно, что у больных БП в обеих группах, различающихся по стадийности процесса, ЭМГ в состоянии покоя характеризуется преимущественно ІІІ типом, что составляет 57,9 ± 2,8 и 87,7 ± 4,1 % случаев соответственно. При функциональных пробах с максимальным разгибанием предплечья у больных І группы со стадией болезни 1,0–2,0 регистрируются усиление залповой активности, на ЭМГ регистрируется смешанный І–ІІІ тип активности в 22,2 ± 2,1 % и значительное уменьшение представленности этого типа до 6,5 ± 0,3 % (р < 0,01) у больных БП при более выраженной (2,5–3,0) стадии заболевания. С другой стороны, наблюдается резкое возрастание представленности залповой активности с 67,4 ± 3,2 до 88,7 ± 4,8 %, что отражает нарушение надсегментарных механизмов регуляции мышечного тонуса на фоне прогрессирующего нейродегенеративного процесса.
Значительный интерес представляют ЭМГ-результаты у родственников больных БП, у которых при неврологическом осмотре были обнаружены субклинические симптомы экстрапирамидной недостаточности. Так, если при ЭМГ покоя определялись в основном І и смешанный (І–ІІІ) типы, что соответствовало 79,5 ± 3,6 и 20,5 ± 1,8 % наблюдений, то при функциональной ЭМГ с максимальным разгибанием и сгибанием предплечья резко возрастала смешанная активность на уровне залповой, что составляло 57,8 ± 2,7 и 62,6 ± 2,9 % соответственно.
Аналогичная тенденция нарастания вольтажа ЭМГ в обследованных группах показана при обследовании родственников больных БП (табл. 4).
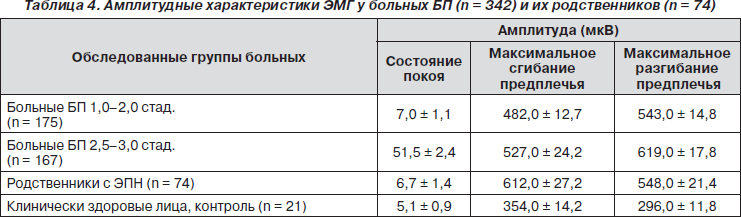
Проведенные ЭМГ-исследования у родственников больных БП дали возможность установить экстрапирамидный тип ЭМГ-активности (залповую активность), свойственный для БП, у большинства обследованных, что дает основание рекомендовать обязательное проведение ЭМГ-диагностики в группах риска, к которым следует относить родственников пробандов. Положительный диагностический ЭМГ-тест можно рассматривать как основание для разработки мер первичной профилактики БП у родственников первой степени родства.
Таким образом, клинико-генеалогический и ЭМГ-анализ обследованных семей указывает на то, что БП наследуется как по аутосомно-доминантному, так и по аутосомно-рецессивному типу. Тот факт, что при БП существуют два типа наследования, свидетельствует о гетерогенности заболевания. По нашим данным, в 76,6 % случаев (23 семьи из 30) болезнь Паркинсона наследовалась по аутосомно-доминантному типу, что, по-видимому, отражает неполную пенетрантность гена (25 %). Низкая пенетрантность доминантного гена указывает на то, что, вероятно, экзогенные факторы (инфекция, травма, стресс) будут играть весьма важную роль в реализации генетически детерминированного патологического признака. Наследование заболевания по аутосомно-рецессивному типу наблюдалось в 23,4 % случаев (7 семей из 30).
Вопрос о причинах гетерогенного типа передачи патологического признака в семьях долгое время оставался открытым. Однако в связи с картированием генов PARKIN’а [28] и развитием учения о молекулярно-генетической основе БП гетерогенный тип наследования БП становится объяснимым.
Клиника проявлений БП, наследуемой по аутосомно-доминантному или аутосомно-рецессивному типу, имеет сходства и различия. При обоих видах наследования БП одинаково часто страдают мужчины и женщины, заболевание начинается после 40 лет. Однако средний возраст больных БП с аутосомно-доминантным типом несколько выше (58,0 ± 2,5 года), чем у пробандов с аутосомно-рецессивным типом наследования (49,0 ± 3,7 года). У больных БП с аутосомно-доминантным типом чаще преобладает дрожательно-ригидная форма заболевания, а при аутосомно-рецессивном типе одинаково часто наблюдаются все формы.
Для профилактики БП в группах риска, к которым относятся родственники больных, очень важно, чтобы больные и их родственники состояли на учете в специализированном центре по экстрапирамидной патологии, при этом важно определять предрасположенность к БП и степень риска заболеть у тех членов семей, у которых обнаружены субклинические и ЭМГ-проявления экстрапирамидной недостаточности. Следует полагать, что эффективная профилактика БП требует своевременного диагностирования доклинической стадии заболевания. Современные клинико-диагностические тесты и ЭМГ позволяют выявить неврологически бессимптомные признаки ЭПН, свидетельствующей о скрытом, клинически компенсированном дефиците активности дофаминергической нигростриатной системы, что может быть связано с дебютом нейродегенеративного процесса.
Можно полагать, что в группу риска должны входить клинически здоровые родственники больных БП, генетически предрасположенные к БП, и особенно те из них, которые находятся в экологически вредных средовых влияниях, использующие фармакологические препараты (нейролептики), длительный прием которых может снижать активность дофаминергической нигростриатной системы. Фактором риска проявления БП у генетически предрасположенных родственников больных может быть сердечно-сосудистая и цереброваскулярная патология. Дебют БП у таких лиц может провоцироваться длительным пребыванием под наркозом при хирургических операциях, острым или хроническим психоэмоциональным стрессом.
1. Polymeropoulos M.N., Higgins G.G., Golbe L.I. Mapping of a gene for Parkinson’s disease to chromosome 4q21-23 // Science. — 1996. — 274. — Р. 1197-1199.
2. Johnson W.I., Hodge S.E., Duvoisin R. Twin studies and the genetics of Parkinson’s disease — a reappraisal // Mov. Disord. — 1990. — 5. — Р. 187-194.
3. Brooks D.J. Functional imaging of Parkinson’s disease: is it possible to detect brain areas for specific symptoms? // J. Neural. Transm., Suppl. — 1999. — Vol. 56. — Р. 139-153.
4. Piccini P., Morrish P.K., Turjanski N. Dopaminergic functuon in familial Parkinson’s disease: a clinical and 18F-dopa positron emission tomography // Ann. Neurol. — 1997. — 41. — Р. 222-229.
5. Nussbaum R., Polymeropoulos M. Genetics of Parkinson’s disease // Hum. Mol. Genet. — 1997. — Vol. 6. — Р. 1687-1691.
6. Spacey S., Wood N.W. The genetics of Parkinson’s disease // Curr. Opin. Neurol. — 1999. — Vol. 12. — Р. 427-432.
7. Elston R.C. Linkage and association // Genet. Epidemiol. — 1998. — Vol. 15. — Р. 565-576.
8. Bonifati V., De Michele G., Lьcking C.B. et al. The parkin gene and its phenotype: Italian PD Genetics Study Group, French PD Genetics Study Group and the European Consortium on genetic susceptibility in Parkinson’s disease // Neurol. Sci. — 2001. — Vol. 20. — Р. 51-52.
9. Farrer M., Kachergus J., Forno L. et al. Comparison of kindreds with familial parkinsonism and synuclein genomic multiplications // Ann. Neurol. — 2004. — 55. — 174-179.
10. Mata I.F., Lockhart P.J., Farrer M.J. Parkin genetics: one model for Parkinson’s disease // Hum. Mol. Genet. — 2004. — 13. — 127-133.
11. Mata I.F., Samii A., Schneer S.Y. et al. Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders // Arch. Neurol. — 2008. — 65. — 379-382.
12. Иллариошкин С.Н. Конформационные болезни мозга. — М.: Янус-К, 2003.
13. Иллариошкин С.Н., Загоровская Т.Б. Иванова-Смоленская И.А., Маркова Е.Д. Генетические аспекты болезни Паркинсона // Неврологический журнал. — 2002. — 5. — 47-52.
14. Шадрина М.И., Багыева Г.Х., Иллариошкин С.Н. и др. Структурные перестройки в гене паркина (PARK2) у больных с паркинсонизмом молодого возраста // Мед. генетика. — 2006. — 12. — 22-26.
15. Klein C., Grunewald A., Hedrich K. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes // Neurology. — 2006. — 66. — 1129-1130.
16. Hedrich K., Djarmati A., Schafer N. et al. DJ-1 (PARK7) mutations in early-onset Parkinson disease // Neurology. — 2004. — 62. — 389-394.
17. Thomas B., Beal M.F. Parkinson’s disease // Hum. Mol. Genet. — 2007. — 16. — R183-R194.
18. Foroud T. LRRK2: both a cause and a risk factor for Parkinson’s disease? // Neurology. — 2005. — 65. — 664-665.
19. Illarioshkin S.N., Shadrina M.I., Slominsky P.A. et al. A common leucine-rich repeat kinase 2 gene mutation in familial and sporadic Parkinson’s disease in Russia // Eur. J. Neurol. — 2007. — 14. — 413-417.
20. Lesage S., Darr A., Tazir M. et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs // New Engl. J. Med. — 2006. — 354. — 422-423.
21. Sidransky E. Gaucher disease: complexity of a «simple» disorder // Mol. Genet. Metab. — 2004. — 83. — 6-15.
22. Lьcking C.B., Dьrr A., Bonifati V. Association between early-onset Parкinson’s disease and mutation in the parkin gene // N. Engl. J. Med. — 2000. — V. 342. — Р. 1560-1567.
23. Periquet M., Fugla T., Myllykangas L. et al. Aggregated a synuclein mediates dopaminergic neurotoxicity in vivo // J. Neurosci. — 2007. — 27. — 3338-3346.
24. Thomas B., Beal M.F. Parkinson’s disease // Hum. Mol. Genet. — 2007. — 16. — R183-R194.
25. Бадалян Л.О., Скворцов И.А. Клиническая электромиография. — М: Медицина, 1986. — 368 с.
26. Крыжановский Г.Н., Карабань И.Н., Магаева С.В., Кучеряну В.Г., Карабань Н.В. Болезнь Паркинсона (этиология, патогенез, клиника, диагностика, лечение, профилактика). — М.: Медицина, 2002. — 355 с.
27. Yavich L., Tanila H., Vepsalainen S., Jakala P. Role of alpha-synuclein in presinaptic dopamine recruitment // J. Neurosci. — 2004. — 24. — 11165-11170