
Журнал «Здоровье ребенка» 5 (32) 2011
Вернуться к номеру
Випадок спадкового генералізованого остеопетрозу в однієї родини
Авторы: Мисько Л.В., Мисько Ю.Л., Юрчишена Е.В., Юрчишен О.М. Хмельницька міська дитяча лікарня Хмельницька обласна дитяча лікарня
Рубрики: Педиатрия/Неонатология
Версия для печати
У статті наведено опис власного клінічного спостереження дітей в одній сім’ї, хворих на спадковий генералізований остеопетроз, клінічний перебіг, особливості діагностики та лікування хворих.
Діти, спадковий генералізований остеопетроз, хвороба Альберс-Шенберга, мармурова хвороба, спадковий злоякісний остеопетроз, генералізований остеосклероз.
Вступ
Генералізований остеопетроз (ОП) (хвороба Альберс-Шенберга, мармурова хвороба (МХ), вроджений злоякісний остеопетроз, генералізований остеосклероз) — спадкове захворювання, що зустрічається рідко та характеризується системним склерозуванням кісток усього скелета, різким зниженням плацдарму медулярного кровотворення та, як наслідок, розвитком вогнищ екстрамедулярного кровотворення у різних паренхіматозних органах [13–15].
За літературними даними [1–3], захворювання спостерігається у популяціях із частотою 1 : 100 000. Незважаючи на те що остеопетроз як самостійна нозологічна форма вперше був описаний німецьким рентгенологом Альберс-Шенбергом у 1904 році, за даними М.О. Ренделя та О.П. Сумакової, до 1935 року у спеціальній літературі було описано лише 46 випадків захворювання. О.В. Лузіна, І.Ш. Акрамов та Б.Д. Гулямов у 1988 році наводять дані про опис у закордонній літературі понад 300 спостережень ОП. Існують свідчення, що до 1976 року в СРСР був описаний лише 41 випадок хвороби Альберс-Шенберга [5, 6].
У теперішній час розрізняють легку автосомно-домінантну, летальну автосомно-рецесивну та помірно-тяжку автосомно-рецесивну форми. За даними О.Н. Конькова (1981), спадковий остеопетроз зустрічається у населення всіх континентів, хворіють особи як чоловічої, так і жіночої статі. Дослідження цих авторів були присвячені вивченню клінічної, рентгенологічної картин та змінам у периферичній крові при даному захворюванні. З метою діагностики мармурової хвороби авторами рекомендовано проводити рентгенологічне дослідження кісткового скелета всім дітям грудного віку з анемією невідомого генезу, гепатоспленомегалією та відставанням у психофізичному розвитку. Багато питань стосовно епідеміології, етіології, патогенезу, генетичних особливостей, вікової варіабельності клінічних проявів ОП до цього часу залишилися не вивченими.
У літературних джерелах відсутні дані щодо факторів ризику при ОП, ранньої діагностики, рекомендацій з профілактики захворювання та організації медичної допомоги сім’ям із високим ризиком народження дітей з ОП чи наявністю у сім’ї хворих дітей. Генералізований ОП до теперішнього часу залишається летальним захворюванням. Крім того, усі хворі з самого раннього віку стають інвалідами. Прояви захворювання практично з народження дитини призводять до великих матеріальних витрат на лікування, догляд, організацію за нею медичного спостереження, виплати пенсії за інвалідністю та інших соціальних пільг. Відомо, що наявність у сім’ї дитини, яка має спадкове захворювання, створює певні проблеми для формування у неї нормального психологічного мікроклімату [9, 12]. Переживання за здоров’я наступних дітей у таких сім’ях загострює проблему планування народження дітей та інколи стає причиною розпаду сім’ї.
Рецесивний летальний остеопетроз є вродженим станом, що обумовлений відсутністю етапу медулярного кровотворення, як у внутрішньоутробному, так і постнатальному періоді розвитку хворої на ОП дитини. У клініці новонароджених з ОП спостерігаються такі основні симптоми: анемія гіпопластичного характеру, екстрамедулярний мієлопроліферативний процес у печінці та селезінці, характерні для екстрамедулярного типу кровотворення зміни у периферичній крові, виражена «бідність», навіть відсутність кістковомозкового кровотворення у картині мієлограми [10, 11].
Остеопетроз є прогресуючим станом із зростанням нормохромної гіпорегенераторної анемії, об’ємів екстрамедулярного кровотворення у печінці, селезінці та інших паренхіматозних органах. При ОП первинно страждає формування кістковомозкового простору як плацдарму кістковомозкового кровотворення, що повністю відповідає уявленню про патогенез ОП, де основна роль належить різкому скороченню та навіть відсутності кісткової резорбції, через що порушується процес формування губчатої кісткової тканини, що відбувається в нормі [16].
Відсутність кістковомозкового простору у внутрішньоутробному періоді розвитку перешкоджає виникненню головного етапу гемопоезу — кістковомозкового, що на кінцевому етапі призводить до порушення формування нормальної гемопоетичної системи хворого ще в період внутрішньоутробного розвитку. При ОП перш за все страждає кістковомозкове кровотворення. У свою чергу, відсутність етапу кістковомозкового кровотворення в організмі хворого на ОП компенсується збереженням навіть після народження ембріонального (у першу чергу печінкового та селезінкового) типів кровотворення та виникненню у подальшому вогнищ екстрамедулярного кровотворення у лімфатичних вузлах та інших паренхіматозних органах. Клінічне збереження ембріонального типу кровотворення у печінці та селезінці у хворих на ОП проявляється гепатоспленомегалією, яка виявляється ще у періоді новонародженості [17].
Клінічно хвороба проявляється переломами кісток, часто зустрічається приглухуватість та гіперпігментація шкірних покривів, відмічається остеомієліт та некроз щелепи, ранній розвиток та прогресуючий перебіг гідроцефального та геморагічного синдромів, постійно присутній міатонічний синдром, прогресування відставання у фізичному, психомоторному та нервово-психічному розвитку [4, 7, 8].
За даними літератури [18], у всіх хворих на ОП виявляються значні зміни у периферичній крові: завжди наявна значна еритроцитопенія та вміст еритроцитів варіює від 1,5 Ч 109/л до 3,5 Ч 109/л. У деяких хворих відмічається циклічність перебігу ОП. У періоді ремісії кількісний уміст еритроцитів у периферичній крові стабілізується та не знижується протягом декількох тижнів і навіть місяців. Показники рівня гемоглобіну в периферичній крові хворих на ОП варіюють у межах від 35–40 до 85–105 г/л залежно від кількості замісних гемотрансфузій, а в періоди ремісії можуть стабілізуватися на рівні 90–95 г/л. У третини хворих наявні ретикулоцитоз та нормобластоз. У всіх хворих спостерігається анізоцитоз та пойкілоцитоз еритроцитів. Значний лейкоцитоз із нейтрофільним зсувом лейкоцитарної формули вліво спостерігається в усіх хворих. Причому у частини хворих наявний гіперлейкоцитоз із умістом формених елементів у межах 35–45 Ч 109/л та вище. Вміст моноцитів у межах 10–15 (рідше — до 20). У більшості хворих у периферичній крові спостерігається нормальний (до 55–60 %) чи дещо підвищений (до 60–65 %) вміст лімфоцитів. Тромбоцитопенія має більш виражену варіабельність — від 45 Ч 109/л до 290 Ч 109/л.
Виникнення компенсаторного екстрамедулярного кровотворення в лімфатичних вузлах клінічно проявляється збільшенням лімфатичних вузлів різних груп. Лімфаденопатія у хворих з віком прогресує, збільшені лімфатичні вузли інколи визначаються візуально, особливо на тлі стоншеної підшкірної клітковини. Як уже відзначалося, екстрамедулярне кровотворення не здатне в повному об’ємі компенсувати потреби організму в формених елементах крові, їх якісний та кількісний склад. У хворих із самого початку розвивається анемія, що, як одна зі складової класичної тріади ОП, супроводжує хворого протягом усього життя, має гіпорегенераторний характер і проявляється стійкою, що погано піддається терапії, еритроцитопенією та гіпогемоглобінемією.
Певне практичне значення мають літературні свідчення про спроби лікування летального остеопетрозу. P.J. Moe та A. Skjaveland (1969) отримали позитивний результат при лікуванні хворих на ОП кортизоном. У дослідженнях L.L. Key (1992, 1995) наводяться дані про досвід лікування хворих на ОП рекомбінантним інтерфероном гамма-1b (interferon gamma-lb), внаслідок чого збільшується резорбція кісток, зменшується трабекулярна кістка та збільшується площа кісткового мозку.
Велику зацікавленість має інформація про ефективні спроби лікування ОП шляхом трансплантації донорського кісткового мозку. Як вважають D.G. Walker (1975) та P.F. Coccia (1980), при трансплантації кісткового мозку центри осифікації можуть бути засіяні остеокластичними джерелами, які дають нові функціонально активні лінії остеокластів. Таким чином, пересадка кісткового мозку є етіотропною терапією при лікуванні спадкового генералізованого остеопетрозу.
На сьогоднішньому етапі найбільш ефективним методом покращення якості та тривалості життя у хворих на ОП є замісна терапія еритроцитарною масою. Але трансфузійна терапія можлива лише в стаціонарних умовах, потребує періодичної госпіталізації дітей та ефективна лише при систематичних повторних дорогих гемотрансфузіях, що призводять до численних ускладнень.
За результатами гістологічного дослідження кісток хворих, які померли від остеопетрозу, у кісткових структурах спостерігаються остеокласти звичайної форми, але в місцях їх локалізації є ознаки резорбції кістки, тобто лакуни не зустрічаються. Кількість остеоцитів менша, ніж звичайно, у частини з них виявляються дистрофічні та некротичні зміни. Патоморфологічне дослідження кісток хворих на ОП підтверджує, що захворювання, яке ми спостерігаємо, дійсно обумовлено мутацією, розміщеною в TCIRG1. Внаслідок мутації у гені TCIRG1 порушений синтез чи структура АТФ-залежного протонного насоса, необхідного для нормального функціонувания остеокластів, а саме їх здатності руйнувати кісткову тканину.
Середня тривалість життя хворих на летальну форму автосомно-рецесивного ОП при посиндромній терапії становить 25,3 ± 3,9 місяця.
Прикладом спадкового генералізованого злоякісного остеопетрозу може бути захворювання дітей в одній сім’ї, яких ми спостерігали на базі Хмельницької обласної дитячої лікарні.
Хворий С. (хлопчик), 05.06.1997 р.н., уперше надійшов до Хмельницької обласної дитячої лікарні (ХОДЛ) 13.10.1997 р. у віці 2,5 місяця. Скарги при надходженні: підвищення температури тіла до 39 °С, геморагічне висипання на нижніх кінцівках. З огляду на наведені скарги дитину спочатку направили до інфекційної лікарні м. Хмельницького з підозрою на менінгококцемію, діагноз не підтвердився, але було виявлено гепатолієнальний синдром та зміни в загальному аналізі крові (анемія, тромбоцитопенія, лейкоцитоз). Серед параклінічних досліджень проведено ультразвукове дослідження органів черевної порожнини (УЗД ОЧП) та діагностичну стернальну пункцію. Через приєднання кишкової інфекції (висіяний золотистий стафілокок) протягом трьох тижнів лікувався в інфекційній лікарні. За час лікування у хлопчика спостерігалася гіпохромна анемія (гемоглобін — 38–56 г/л), тромбоцитопенія (50–70 • 109/л), лейкоцитоз (23–28 • 109/л), гепатолієнальний синдром. При надходженні до ХОДЛ у дитини відзначалася лімфаденопатія, збільшення печінки +5 см, селезінки +6 см нижче краю реберної дуги.
Проведено дообстеження дитини: загальний аналіз крові, біохімічний аналіз крові, стернальна пункція з мієлограмою, УЗД ОЧП.
Загальний аналіз крові: 14.10.1997 р. — гемоглобін (Hb) — 73 г/л, еритроцити — 3,01 • 1012/л, кольоровий показник (КП) — 0,73, лейкоцити — 39 • 109/л, тромбоцити — 50 • 109/л, ретикулоцити — 162 ‰, промієлоцити — 1 %, мієлоцити — 7 %, юні — 7 %, паличкоядерні (п/я) — 15 %, сегментоядерні (с/я) — 16 %, лімфоцити — 32 %, моноцити — 9 %, базофільні нормобласти — 2, поліхроматофільні нормобласти — 19, оксифільні нормобласти — 18 на 100 лейкоцитів, швидкість осідання еритроцитів (ШОЕ) — 23 мм/год.
Біохімічний аналіз крові: 14.10.1997 р. — загальний білок — 65,4 г/л, альбуміни — 44,9 г/л, глобуліни — 20,5 г/л, аспартатамінотрансфераза (АсАт) — 0,85 ммоль/л, аланінамінотрансфераза (АлАт) — 0,33 ммоль/л, сечовина — 3,2 ммоль/л, білірубін — 13,7 ммоль/л.
Мієлограма: недиференційовані бластні клітини — 9 %; нейтрофільні промієлоцити — 0,2 %, мієлоцити — 7 %, метамієлоцити — 3,4 %, п/я — 10,6 %, с/я — 22,3 %, еозинофіли — 4,8 %, базофіли — 0,8 %, лімфоцити — 13,8 %, моноцити — 0,8 %, плазматичні клітини — 0,4 %, еритробласти — 1,6 %. Нормобласти: базофільні — 5,8 %, поліхроматофільні — 23 %, оксифільні — 4,6 %. Кількість мієлокаріоцитів — 43 • 109/л, кількість мегакаріоцитів — 0,006 • 109/л, лейкоеритробластичне співвідношення — 1,9 : 1. Індекс дозрівання нейтрофілів — 0,3, індекс дозрівання еритробластів — 0,8. Кістковомозкове кровотворення пригнічено. Відносне подразнення еритроїдного ростка. Кровотворення за нормобластним типом. Зустрічаються поліхроматофільні та оксифільні нормобласти з «мегалобластними» ядрами. Анізоцитоз (+), пойкілоцитоз (+), поліхроматофілія (+). Мітози — 2–3 : 100 клітин. Мегакаріоцитарний росток пригнічений. Індекс дозрівання нейтрофілів низький.
УЗД ОЧП. Печінка: передньозадній розмір правої частки 8,4 см, структура однорідна. Жовчний міхур: овально-продовгуватої форми. Підшлункова залоза: без особливостей. Селезінка: 8,9 ґ 2,8 см, структура однорідна. Нирки: без особливостей.
З огляду на анамнез захворювання, клінічні та лабораторні дані запідозрено діагноз хронічного мієлобластного лейкозу, тому дитина була направлена на консультацію в «ОХМАТДИТ» (м. Київ) на консультацію до гематолога, але батьки забрали дитину зі стаціонарного лікування додому. Протягом двох років лікувалися нетрадиційними методами. Повторно батьки з дитиною звернулися по медичну допомогу 15.09.1999 р. через різку блідість шкірних покривів та слизових оболонок, виражений геморагічний синдром на шкірі, збільшення в об’ємі живота та зміни в загальному аналізі крові.
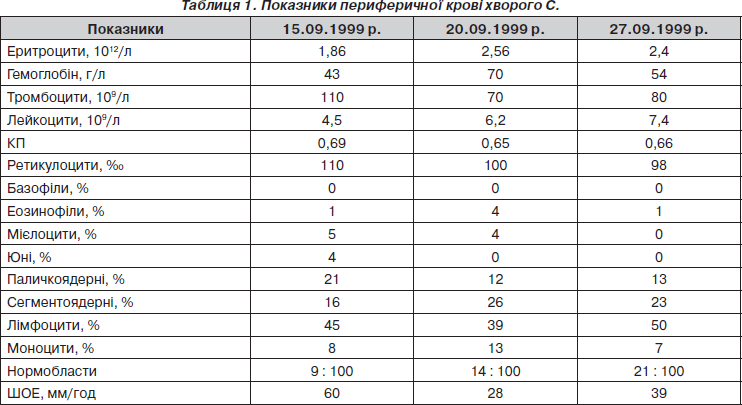
У гемограмі відзначалися анемія, тромбоцитопенія, ретикулоцитоз, нормобласти, анізоцитоз, пойкілоцитоз (+), поліхромазія (++) (табл. 1).
Біохімічне дослідженні крові 16.09.1999 р.: загальний білок — 38 г/л, альбуміни — 21,3 г/л, глобуліни — 16,7 г/л, АсАт — 0,45 ммоль/л, АлАт — 0,15 ммоль/л, сечовина — 4,3 ммоль/л, білірубін — 20,5 ммоль/л.
У мієлограмі від 16.09.1999 р.: недиференційовані бластні клітини — 3 %; нейтрофільні промієлоцити — 2,5 %, мієлоцити — 6,5 %, метамієлоцити — 5,5 %, п/я — 11,5 %, с/я — 19,5 %, еозинофіли — 0 %, базофіли — 0 %, лімфоцити — 18,5 %, моноцити — 2 %, еритробласти — 1,5 %. Нормобласти: базофільні — 7 %, поліхроматофільні — 16 %, оксифільні — 6,5 %. Кількість мієлокаріоцитів — 43 • 109/л, кількість мегакаріоцитів — 0,006 • 109/л, лейкоеритробластичне співвідношення — 2,2 : 1. Індекс дозрівання нейтрофілів — 0,46, індекс дозрівання еритробластів — 0,7. Кістковомозкове кровотворення пригнічено, є ознаки подразнення еритроїдного ростка. Мегакаріоцитарний росток пригнічений. Індекс дозрівання нейтрофілів низький.
Обстежений на токсоплазмоз та цитомегаловірусну інфекцію (методом ІФА). Виявлено IgG до токсоплазми в концентрації 300 МЕ/мл (за нормою 0,01–9,99), на цитомегаловірусну інфекцію — результат негативний.
УЗД ОЧП. Печінка: +7,0 см, підвищеної ехогенності, печінкова вена — 0,8 см. Жовчний міхур: овально-продовгуватої форми. Підшлункова залоза: без особливостей. Селезінка: 127 ґ 57 мм, структура однорідна. Нирки: права — 7,4 ґ 3,5 см, ліва — 6,9 ґ 2,8 см, товщина паренхіми — 12 мм.
Нейросонограма (НСГ): зміщення центральних ехоструктур немає, базальний рисунок середнього мозку, базальних цистерн без особливостей, відмічається розширення його тіла, вентрикулодилатація бокових шлуночків, розширення її щілини.
Консультації. Окуліст: субатрофія зорових нервів обох очей. Інфекціоніст: хронічний токсоплазмоз.
З огляду на анамнез, клінічний перебіг захворювання та дані лабораторного обстеження дитина направлена на консультацію в Інститут педіатрії, акушерства та гінекології (м. Київ), де встановлено клінічний діагноз: спадковий рецесивний остеосклероз (мармурова хвороба); мієлодиспластичний синдром; синдром гіперспленізму; субатрофія зорових нервів обох очей; хронічний токсоплазмоз.
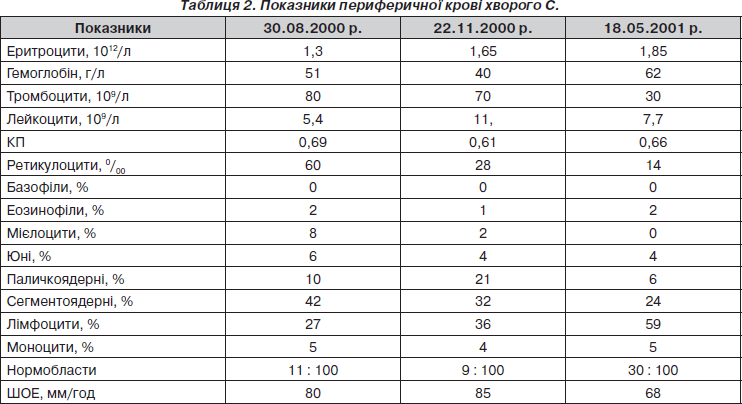
Перебіг захворювання ускладнився флегмоною нижньої щелепи на тлі вираженої анемії, тромбоцитопенії (табл. 2).
В останні місяці життя у дитини розвинувся остеомієліт нижньої щелепи зліва, що у подальшому ускладнився сепсисом з летальним кінцем.
У цій родині народилося дві дівчинки (2002 та 2008 року народження), які є абсолютно здоровими. Четверта дитина (хлопчик В.) народилася 09.02.2011 р. доношеною з масою тіла 3650 грамів. Надійшов на лікування до ХОДЛ зі скаргами на блювоту, відставання у масі тіла, збільшення в об’ємі живота та зміни в загальному аналізі крові: анемія, тромбоцитопенія, лейкоцитоз. При обстеженні у дитини відзначалися: блідість шкірних покривів та слизових оболонок, відставання у масі тіла на 25 %, збільшення печінки +3 см, селезінки +2,5 см, систолічний шум над серцевою ділянкою.
Параклінічне обстеження дитини включало: загальний аналіз крові, біохімічний аналіз крові, стернальну пункцію з мієлограмою, УЗД ОЧП, рентгенографію кистей та кісток нижніх кінцівок, НСГ.
Загальний аналіз крові від 26.04.2011 р.: Hb — 71 г/л, еритроцити — 3,04 • 1012/л, КП — 0,7, лейкоцити — 21,2 • 109/л, тромбоцити — 60–70 • 109/л, ретикулоцити — 68 ‰, промієлоцити — 10 %, мієлоцити — 3 %, юні — 6 %, п/я — 10 %, с/я — 24 %, лімфоцити — 38 %, моноцити — 4 %, ШОЕ — 30 мм/год, поліхроматофільні нормобласти — 5 : 100.
Мієлограма: недиференційовані бластні клітини — 3 %, нейтрофільні промієлоцити — 4 %, мієлоцити — 7 %, метамієлоцити — 4 %, п/я — 6 %, с/я — 16 %, еозинофіли — 3 %, лімфоцити — 40 %, моноцити — 8 %, еритробласти — 5 %. Нормобласти: базофільні — 2 %, поліхроматофільні — 2 %, оксифільні — 4 %. Кількість мієлокаріоцитів — 40 • 109/л, кількість мегакаріоцитів — 0,007 • 109/л, лейкоеритробластичне співвідношення — 1,9 : 1. Індекс дозрівання нейтрофілів — 0,3, індекс дозрівання еритробластів — 0,8. Кістковомозкове кровотворення пригнічене. Відносне подразнення еритроїдного ростка. Кровотворення за нормобласним типом. Анізоцитоз (+), пойкілоцитоз (+), поліхроматофілія (+). Мітози 2–3 : 100 клітин. Мегакаріоцитарний росток пригнічений. Індекс дозрівання нейтрофілів низький.
Біохімічний аналіз крові: загальний білок — 65,4 г/л, альбуміни — 44,9 г/л, глобуліни — 20,5 г/л, АсАт — 0,85 ммоль/л, АлАт — 0,33 ммоль/л, сечовина — 3,2 ммоль/л, білірубін — 13,7 ммоль/л, сироваткове залізо — 47 мкмоль/л, загальнозв’язуюча здатність — 83 мкмоль/л, прихована загальнозв’язуюча здатність — 36 мкмоль/л.
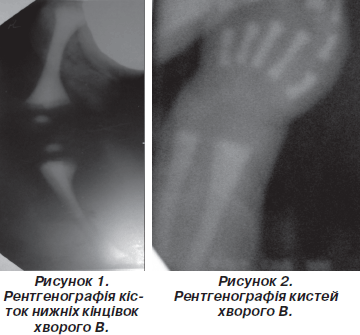
Рентгенографія кистей та кісток нижніх кінцівок: остеосклероз, остеопетроз усіх кісток, бутелеподібна деформація метафізів (риc. 1, 2).
НСГ: незначне розширення міжоболонкового та підоболонкового простору по контекстній поверхні півкуль мозку.
УЗД ОЧП. Печінка: передньозадній розмір правої частки — 6,8 см, структура зерниста, ехогенність помірно підвищена. Жовчний міхур: овально-продовгуватої форми. Підшлункова залоза: розміри розширені, підвищеної ехогенності. Селезінка: 7,9 ґ 2,4 см, структура однорідна, помірно збільшена. Нирки: без особливостей.
Консультації. Окуліст: часткова атрофія дисків зорових нервів обох очей. Генетик: спадковий рецесивний остеосклероз (мармурова хвороба). Травматолог: спадковий рецесивний остеосклероз.
На даному етапі хворий отримує замісну трансфузію еритроцитарної маси.
Профілактика генералізованого остеопетрозу у дітей включає систему заходів профілактики нових випадків остеопетрозу:
— алгоритм діагностики та профілактики народження дітей зі спадковим остеопетрозом;
— Республіканський реєстр спадкового остеопетрозу;
— система медичної допомоги сім’ям з високим ризиком народження дітей з летальним остеопетрозом;
— масовий ультразвуковий скринінг вагітних жінок у терміні вагітності 21–22 тижні гестації сприяє запобіганню народження дітей зі спадковим остеопетрозом;
— скринінг на гетерозиготне носійство мутації в гені TCIRG1 у певних груп населення для цього захворювання зміг би попередити народження дітей із цією патологією.
Висновки
1. Спадковий генералізований остеопетроз є системним захворюванням, при якому в патологічний процес утягнуті всі без винятку кістки краніофасціального, апендикулярного та осьового скелета, що має принципове значення для диференційної діагностики остеопетрозу з іншими захворюваннями, що супроводжуються склерозуванням або надмірною осифікацією кісткового скелета.
2. Оптимізація прогнозу життя хворих на спадковий остеопетроз дітей на сучасному етапі перш за все пов’язана з трансплантацією кісткового мозку, що сприяє відновленню кісткової резорбції та необхідна для формування в кістках кістковомозкових порожнин як плацдарму кістковомозкового кровотворення.
3. Трансплантація кісткового мозку не знешкоджує нейросенсорні порушення, що вже існують, тому мієлотрансплантація повинна проводитися в максимально ранні строки життя хворих дітей до настання «тунельного» защемлення черепно-мозкових нервів.
4. Найбільш успішною є трансплантація кісткового мозку, коли донорами кісткового мозку для реципієнтів є їх HLA-MLA-ідентичні сібси, але це проблемно для первістків.
5. Альтернативою дорогої, не завжди успішної та не знешкоджуючої існуючих нейросенсорних порушень мієлотрансплантації повинна стати профілактика народження дітей зі спадковим генералізованим остеопетрозом, тобто ефективні методи пренатальної діагностики захворювання, що сприяють запобіганню народження дітей з цією патологією.
1. Антимоний Р.Г., Сысуев Л.А. Случай мраморной болезни у ребенка // Вестник рентгенологии и радиологии. — 1980. — № 3. — С. 71-72.
2. Бобырева С.Б., Ковалев М.Ф. Семейный случай мраморной болезни // Педиатрия. — 1980. — № 2. — С. 58.
3. Вихрева Е.А., Бычковский В.П., Хейфец С.Н. Случай семейной формы мраморной болезни (синдрома Альберс-Шенберга) // Педиатрия. — 1984. — № 1. — С. 67-68.
4. Гинтер Е.К., Кириллов А.Г. Ультразвуковая пренатальная диагностика остеопетроза // Информационное письмо / Чувашский университет. — Чебоксары, 2004. — 8 с.
5. Журавлева И.В., Еникеев Р.Г., Беляев С.Е. Два случая врожденной мраморной болезни // Вопросы охраны материнства и детства. — 1980. — № 11. — С. 71-72.
6. Зейналова А.А., Дадашева Т.С., Абраханова X.Н. Врожденная мраморная болезнь и несфероцитарная гемолитическая анемия, обусловленная дефицитом Г-6-ФДГ в эритроцитах у мальчика в возрасте 2 лет // Педиатрия. — 1981. — № 6. — С. 72-73.
7. Кириллов А.Г., Гинтер Е.К. Опыт пренаталыюй диагностики врожденного остеопетроза // Медицинская генетика. — 2004. — Т. 3, № 10. — С. 490-492.
8. Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Остеопетроз рецессивный // Наследственные синдромы и медико-генетическое консультирование. — М.: Практика, 1996. — С. 205-206.
9. Коньков А.Н. Случай мраморной болезни в сочетании с соматическим инфантилизмом и выраженной анемией // Проблемы гематологии и переливания крови. — 1981. — № 6. — С. 48-50.
10. Марцишевская Р.Д., Куклина М.А. Анемия как одна из масок мраморной болезни // Тезисы докладов III Всесоюзного съезда врачей-лаборантов. — Ч. 2. — М., 1985. — С. 112-114.
11. Некачалов В.В. Патология костей и суставов: Руководство. — СПб.: Сотис, 2000. — 288 с.
12. Asou Y., Rittling S.R., Yoshitake H., Tsuji K., Shinomiya K., Nifuji A., Denhardt D.T., Noda M. Osteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone // Endocrinology. — 2001. — Vol. 142. — P. 1325.
13. Boskey A., Spevak L., Tan M., Doty S.B., Butler W.T. Dentin sialoprotein (DSP) has limited effects on in vitro apatite formation and growth // Calcif. Tissue Int. — 2000. — Vol. 67. — P. 472-478.
14. Corbacioglu S., Schulz A. Lebervenen — verschlessenkrankung im Kindersalter // Monatsschr Kindcrheilkd. — 2003. — Vol. 151. — Р. 174-181.
15. Chalhoub N. Grey-lethal mutation induces severe malignant futo-somal recessive Osteopetrosis in mouse and human // Nat. Med. — 2003. — Vol. 9(4). — Р. 399-406.
16. Denhardt D., Giachelli C., Rittling R. Role of osteopontin in cellular signaling and toxicant injury // Annu. Rev. Pharmacol. Toxicol. — 2001. — Vol. 41. — P. 723-749.
17. Schulz A.S., Classen C.F., Mihatsch W.A., Sigl-Kraetzig M., Wiesneth M., Debatin K.M., Friedrich W., Muller S.M. HLA-haploidentical blood progenitor cіll transplantation in osteopetrosis // Blood. — 2002. — Vol. 99. — Р. 3458-3460.
18. Teitelbaum S. Genetic regulation of osteoclast development and funetion // Nat. Rev. Genet. — 2003. — Vol. 4(8). — Р. 638-649.