
Журнал «Здоровье ребенка» 5 (56) 2014
Вернуться к номеру
Первичный гемофагоцитарный лимфогистиоцитоз у новорожденного ребенка
Авторы: Кожемяка А.И., Клименко В.А., Кандыба В.П. - Харьковский национальный медицинский университет; Пионтковская О.В., Халтурина Т.А., Постников А.В., Перхун М.И., Зоря О.А. - КУЗ «ОДКБ № 1», г. Харьков
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
В статье представлены данные клинического наблюдения гемофагоцитарного лимфогистиоцитоза у новорожденного ребенка, осложненного спонтанным разрывом селезенки. Обсуждены вопросы диагностики согласно рекомендациям Международного общества по изучению гистиоцитарных заболеваний (Histiocyte Society, 2004).
У статті наведено данi клінічного спостереження гемофагоцитарного лімфогістіоцитозу в новонародженої дитини, ускладненого спонтанним розривом селезінки. Обговорені питання діагностики згідно з рекомендаціями Міжнародного товариства з вивчення гістіоцитарних захворювань (Histiocyte Society, 2004).
The article presents the data of clinical observation of hemophagocytic lymphohistiocytosis in newborn child, complicated with spontaneous splenic rupture. Issues of diagnosis according to the recommendations of the International society for the study of histiocytic diseases (Histiocyte Society, 2004).
дети, гемофагоцитарный лимфогистиоцитоз, диагностика.
діти, гемофагоцитарний лімфогістіоцитоз, діагностика.
children, hemophagocytic lymphohistiocytosis, diagnosis.
Статья опубликована на с. 105-108
Прижизненная диагностика гемофагоцитарного лимфогистиоцитоза (ГЛГ) представляет большую редкость. Впервые это заболевание описано J. Farquhar и A. Chaireaux в 1952 г. у двух детей одной семьи (мальчика и девочки) в возрасте 9 недель с признаками гепатоспленомегалии, панцитопении и лимфоидных «размазанных» клеток в периферической крови. В дальнейшем появились сообщения McMahon (1963) и Bell (1968), в которых описываются случаи заболевания у детей в возрасте от 9 недель до 10 месяцев с выраженной анемией, лихорадкой, резко выраженной гепатоспленомегалией и незначительной лимфаденопатией. В периферической крови этих детей наблюдались гемолитическая анемия с полихромазией, ретикулоцитозом, нормобластами, умеренная нейтропения и тромбоцитопения. В миелограмме отмечены эритроидная гиперплазия с уменьшением миелоидного и мегакариоцитарного ростков, большие базофильные эритрофагоцитирующие гистиоциты, поглощающие нормобласты на всех стадиях созревания.
Согласно современной классификации главные формы детского гистиоцитоза сгруппированы в три класса:
I класс:
— гистиоцитоз из клеток Лангерганса;
— ювенильная ксантогранулема;
II класс:
— гемофагоцитарный лимфогистиоцитоз;
— синус-гистиоцитоз с массивной лимфаденопатией;
III класс:
— злокачественные опухоли кроветворения;
— моноцитарная лейкопения;
— злокачественные гистиоцитозы;
— опухоли, связанные с патологией макрофагов (диссеминированные или локализированные).
Гемофагоцитарный лимфогистиоцитоз относится ко II классу гистиоцитозов. Это аутосомно-рецессивное заболевание. Частота встречаемости (по зарубежным сообщениям) составляет 1 : 50 000 новорожденных. Считают, что причиной ГЛГ являются нарушения регуляции клеточного иммунитета с нарушением цитотоксической активности Т-лимфоцитов и функции натуральных киллеров (NK-клеток).
При ГЛГ происходит персистирующая избыточная активация антигенпрезентирующих клеток (макрофагов) цитокинами, которые продуцируются СD8+ Т-клетками, главным образом IFN-y, IL-2, IL-12.
Установлено, что важная роль, наряду с другими факторами, в регуляции цитотоксического действия лимфоцитов принадлежит белку перфорину. Являясь одним из эффекторов цитотоксичности лимфоцитов, перфорин способствует образованию пор в мембране клеток-мишеней и совместно с гранзимом В (семейство сериновых протеаз) индуцирует запрограммированную смерть клеток (апоптоз). Таким образом, перфорин реализует цитотоксическое действие лимфоцитов при врожденном и адаптивном иммунитете.
Дефицит перфорина, связанный с миссенс-мутацией его кодирующего гена, обусловлен выработкой неполных либо нефункциональных полипептидов на рибосомах.
При семейном ГЛГ наблюдаются различные варианты мутации гена перфорина. В молекулярно-генетических исследованиях последнего времени установлена локализация четырех генов, мутации которых приводят к ГЛГ (хромосомы 6, 10, 17, 19). По данным российских генетиков, наиболее частые миссенс-мутации гена перфорина при семейном ГЛГ установлены в 17-й хромосоме.
Диагностика
Диагностика заболевания представляет определенные трудности. Клинически ГЛГ манифестирует упорной лихорадкой, спленомегалией, гепатомегалией, иногда — кожной сыпью, желтухой, отеками, кровоточивостью. В клиническом анализе крови: снижение количества эритроцитов, гемоглобина, тромбоцитов и лейкоцитов.
Диагностика ГЛГ должна базироваться на критериях, предложенных международным обществом по изучению гистиоцитарных заболеваний (Histiocyte Society, 2004):
— лихорадка ≥ 38,5 °С > 7 дней;
— спленомегалия > 3 см ниже края реберной дуги;
— цитопения ≥ в 2 линиях;
— гемоглобин < 90 г/л, тромбоциты < 100 • 109, нейтрофилы < 1 • 109;
— гипертриглицеридемия ≥ 2,0 ммоль/л, гипофибриногенемия ≤ 1,5 г/л;
— ферритин ≥ 500 мкг/л;
— СD25 ≥ 2500 Ед/л;
— cнижение активности NK-клеток;
— гемофагоцитоз в костном мозге, лимфоузлах.
Для установления диагноза ГЛГ необходимо наличие 5 из этих критериев. Диагноз первичного семейного (генетически детерминированного) ГЛГ требует подтверждения молекулярно-генетическим анализом.
В лечении ГЛГ используется комбинированная иммуносупрессивная химиотерапия с включением дексаметазона, этопозида, циклоспорина А (протокол лечения ГЛГ-2004). Всем пациентам с доказанным первичным гемофагоцитарным лимфогистиоцитозом показана аллогенная трансплантация стволовых клеток крови.
Приводим собственное клиническое наблюдение ГЛГ семейного типа у новорожденного ребенка.
Выписка из истории болезни 19214/680 ребенка Г.
Ребенок Г., 9 дней, доставлен МСП из района. Жалобы матери: повышение температуры до 39 °С, выраженное беспокойство. Болен сутки. Была вызвана машина скорой помощи, и ребенок доставлен в ОДКБ № 1 г. Харькова.
Анамнез жизни
Ребенок родился от II беременности, в сроке гестации 37 недель. Первая беременность закончилась рождением доношенного ребенка, который умер в возрасте 1 месяца 8 дней. Патологоанатомический диагноз: врожденный лейкоз. Данная беременность протекала на фоне урогенитальной инфекции (уреаплазмоз, гарднереллез), периодических герпетических высыпаний на губах у матери. Роды самостоятельные, родился мальчик с массой 2940 г, длина тела — 48 см, окружность головы — 34 см, оценка по шкале Апгар — 8–9 баллов. Приложен к груди в родзале. За время пребывания в перинатальном центре состояние оставалось удовлетворительным. Были отмечены экхимозы на коже предплечий, кистей и стоп, выявлено увеличение размеров печени на 2 см и селезенки на 1 см.
Группа крови матери: O(I) Rh-положительный. Группа крови ребенка: А(II) Rh-положительный. Билирубин пуповинной крови — 51,8 мкмоль/л. Почасовой прирост — 7,0 мкмоль/л, в дальнейшем через 18 ч — 91,2 мкмоль/л, на 2-е сутки — 158 мкмоль/л (непрямой 150 мкмоль/л). На 4-е сутки — 167 мкмоль/л, на 6-е — 135 мкмоль/л. Реакция Кумбса отрицательная. Был установлен диагноз ГБН по системе АВО, желтушная форма, среднетяжелое течение. Проведена фототерапия.
В клинических анализах крови в 1-е сутки: эритроциты — 5,4 • 1012/л, Hb — 189 г/л, Нt — 54 %, тромбоциты — 359 • 109/л, лейкоциты — 12,5 • 109/л, нейтрофилы — 26 %, лимфоциты — 24 %. Глюкоза крови, СРБ, общий белок, уровень альбумина, мочевой кислоты, холестерина, амилазы, ЛДГ, ЩФ, креатинина, магния, Са, АЛТ, АСТ в пределах возрастной нормы.
В динамике пребывания в перинатальном центре желтуха уменьшилась, анализы крови оставались в пределах нормы.
Учитывая семейный анамнез, наличие у ребенка желтухи и геморрагического синдрома, провели консилиум с участием гематолога, невропатолога и генетика. Убедительных данных в пользу врожденных заболеваний крови не установлено. Выписан домой на 6-е сутки жизни в удовлетворительном состоянии.
/107/107.jpg)
При поступлении в отделение интенсивной терапии и реанимации новорожденных состояние ребенка расценено как тяжелое. Выражены признаки угнетения ЦНС, бледность кожных покровов с иктеричным оттенком, микроциркуляторные нарушения, температура тела 38 °С. Функция внешнего дыхания и гемодинамика стабильны. Живот вздут, отечность передней брюшной стенки. Пальпаторно выявлена гепатоспленомегалия (печень увеличена на 3 см, селезенка — на 6 см ниже края реберной дуги), увеличение шейных, подмышечных и паховых лимфатических узлов до 1 см в диаметре. В клиническом анализе крови: эритроциты — 3,0 • 1012/л, Hb — 90 г/л, Нt — 27 %, тромбоциты — 35 • 109/л, лейкоциты — 3,5 • 109/л, нейтрофилы — 20 %, лимфоциты — 79 %, моноциты — 1 %.
В динамике заболевания прогрессировала анемия (Hb 60–64 г/л), лейкоцитопения (2,4–3,9) • 109/л, лимфоцитоз 82–93 %, тромбоцитопения (23–25) • 109/л.
Ребенок обследован на TORCH-комплекс, результаты исследований отрицательные. У матери в крови обнаружены IgG и IgM к хламидиям, у ребенка обнаружены материнские IgG к хламидиям и герпесу.
В коагулограмме выявлены гипокоагуляция, удлинение ВСК до 9 мин, снижение протромбинового индекса до 35–40 %, снижение фибриногена до 0,80 г/л, в связи с чем требовались трансфузии замороженной плазмы.
Уровень билирубина при поступлении — 186,6 мкмоль/л (преимущественно неконъюгированный). АЛТ — 3,29 мкмоль/л/ч, АСТ — 3,43 мкмоль/л/ч, уровень сахара — 5,4 ммоль/л.
По данным УЗИ, выражена гепатоспленомегалия, наличие жидкости в брюшной полости, персистирующее фетальное кровообращение (функционирующее овальное отверстие).
На Rö-грамме органов грудной клетки выявлена тимомегалия II ст. В динамике развилась полисегментарная пневмония.
На основании вышеперечисленных жалоб, семейного анамнеза и дополнительных исследований выставлен предположительный диагноз: врожденный лейкоз? Внутриутробная инфекция. Анемия. Тимомегалия II ст. Гипоксически-метаболическая энцефалопатия (синдром нарушения мышечного тонуса, висцеральных дисфункций).
Проведена пункция костного мозга. Миелограмма выполнена в лаборатории клиники «ОХМАТДЕТ» (г. Киев). В препарате костного мозга выявлены небольшое количество клеточных элементов, раздражение миелокариоцитарного ростка, сужение миелоидного и эритроцитарного ростков, увеличение бластных клеток до 10,4 %, единичные клетки типа гистиоцитов (3 %), отмечаются увеличенное количество моноцитов с выраженной плазматизацией и вакуолизированной цитоплазмой, широкоплазменные лимфоциты с плазматизацией цитоплазмы. Мегакариоциты — небольшое количество, иные без зрелых тромбоцитов.
Рекомендовано проводить контроль гемограммы и лейкоцитограммы в динамике, повторное исследование миелограммы через 10–14 дней. Проводить дифференциальную диагностику между врожденным лейкозом и первичным (семейным) гемофагоцитарным лимфогистиоцитозом.
В связи с этим был созван консилиум с привлечением специалистов: главного педиатра МЗ Украины, зав. гематологическим отделением клиники «ОХМАТДЕТ», областного гематолога (г. Харьков), доцента кафедры медицинской генетики Харьковского национального медицинского университета, зав. кафедрой детской хирургии, сотрудников педиатрических кафедр Харьковского национального медицинского университета, зав. отделением реанимации и интенсивной терапии, лечащего врача. Анализ данных обследования больного (семейный анамнез анемии, лейкоцитопении, тромбоцитопении), результатов миелограммы и исследований медико-генетического центра (повышенное содержание триглицеридов, повышенное содержание ферритина более чем в 10 раз) позволил поставить диагноз гемофагоцитарного лимфогистиоцитоза семейного типа.
В связи с верификацией диагноза проведена коррекция терапии с включением иммунозаместительной терапии дексаметазоном с последующим переходом лечения согласно протоколу ГЛГ-2004.
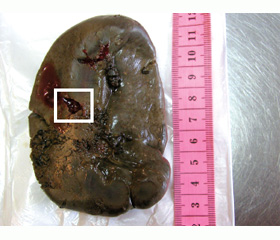
За время пребывания в стационаре состояние ребенка прогрессивно ухудшалось. На 7-й день на фоне резко выраженного тромбогеморрагического синдрома развился геморрагический шок с признаками внутрибрюшного кровотечения. Диагностирован спонтанный разрыв селезенки. Селезенка размером 10 x 6 x 3 см, вес 97 г, капсула утолщенная, сероватая с желтоватым оттенком. По внутренней поверхности селезенки около зоны ворот в средней части имеется поперечный щелевидный разрыв капсулы длиной 1,5 см. Ткань селезенки мягко-эластична, на разрезе однородная, темно-красная. Пульпа селезенки образует умеренный густой кровяной соскоб. Проведена спленэктомия.
Микроскопически определяется густая инфильтрация ткани селезенки активированными макрофагами с признаками активного эритрофагоцитоза, преимущественно в зоне синусов. В межсинусовой ткани признаки лимфоцитоза с гемофагоцитозом клеток. Характер морфологических изменений соответствует гемофагоцитарному лимфогистиоцитозу.
В послеоперационном периоде состояние ребенка оставалось крайне тяжелым, развилась ОПН (уровень мочевины — 16,7 ммоль/л, креатинина — 0,200 ммоль/л, периферические отеки, протеинурия, олигурия).
При нарастании полиорганной недостаточности и некупируемого тромбогеморрагического синдрома наступила остановка эффективного кровообращения, проводимые реанимационные мероприятия были неэффективны. Констатирована смерть ребенка.
Заключительный диагноз
Основной: гемофагоцитарный лимфогистиоцитоз семейного типа, С96.7.
Осложнения: спонтанный разрыв селезенки. Состояние после операции спленэктомии. Синдром полиорганной недостаточности (недостаточность печеночная, почечная, кардиореспираторная, геморрагический синдром). Гипоксически-метаболическое поражение ЦНС (синдром угнетения мышечного тонуса, висцеральных дисфункций).
Сопутствующий диагноз: двусторонняя полисегментарная пневмония. Персистирование фетального кровообращения (Ф00). Вторичная кардиомиопатия. Тимомегалия II ст. Водянка правого яичка.
Заключение патологоанатомического вскрытия ребенка Г., 1 мес.
Патологоанатомический диагноз
Основной: гемофагоцитарный лимфогистиоцитоз — С96.7. Причина смерти: полиорганная недостаточность.
Микроскопически: гемофагоцитарно-лимфогистиоцитарная инфильтрация селезенки, печени, надпочечников, стенки желудка, почек. Очаговая лимфогистиоцитарная инфильтрация мягкой мозговой оболочки, части лимфоузлов. Миелодиспластическое состояние клеточного костного мозга. Гепатоспленомегалия (вес печени — 260 г, селезенки — 97 г).
Осложнения: спонтанный разрыв селезенки с внутрибрюшным кровотечением. Состояние после верхнесрединной лапаротомии, спленэктомии.
Геморрагический синдром: массивные внутрилегочные кровоизлияния, обширные кровоизлияния в слизистой оболочке желудка; перисинусовые пятнистые кровоизлияния в твердой мозговой оболочке.
Двусторонняя очаговая некротическая пневмония. Отек-набухание головного мозга. Двусторонний гидроторакс, асцит. Гипотрофия.
Представленное клиническое наблюдение поможет практикующим врачам более глубоко ознакомиться с данной патологией у детей раннего возраста.
1. Farguhar J.W. Familial haemphagocytic reticulosis / J.W. Farguhar, A.E. Chaireaux // Arch. Dis. Childh. — 1952. — № 27. — P. 519.
2. MacMahon H.E. Familial erytrophagocytic limphohistiocytosis / H.E. MacMahon, M. Bedizel, C.A. Ellis // Pediatrics. — 1963. — № 32. — P. 868.
3. Bell R.I.M. Familial haemophagocytic reticulosis / R.I.M. Bell, A.I.E. Bratied, N.D. Barnes // Arch. Dis. Childh. — 1968. — № 43. — P. 601.
4. Caiz J. Лимфоретикулярные болезни / J. Caiz, B.W. Yancock, J. Hanry, A.M. Ward: Пер. с англ. — М.: Медицина, 1980. — С. 174–177.
5. Blennow Y. Haemophagocytic reticulosis. A state of chimerism? / Y. Blennow, B. Berg, L. Brandit еt al. // Arch. Dis. Childh. — 1979. — № 2. — P. 1178.
6. Nezelof C. La Limphocytose familiale / Nezelof C., Eliachar E. // Nouv. Rev. Franc. Hemat. — 1973. — № 13. — P. 319–338.
7. Гаднер Х. Гистиоцитозы детского возраста / Под ред. Х. Гаднера, А.Г. Румянцева. — М.; Вена: МаксПресс, 2005.
8. Новикова Г.А. Гемофагоцитарные лимфогистиоцитозы у детей: Автореф. дис... канд. мед. наук: спец. 14.01.08 «педиатрия» / Г.А. Новикова. — М., 1999. — 20 с.
9. Милованова Н.В. Молекулярно-генетическая природа первичных гемофагоцитарных лимфогистиоцитозов в России: Автореф. дис... канд. мед. наук: спец. 03.02.07 «генетика», 14.01.21 «гематология и переливание крови» / Н.В. Милованова. — М., 2011. — 20 с.
1. Farguhar J.W., Chaireaux A.E. Familial haemphagocytic reticulosis. Arch. Dis. Childh 1952; 27: 519.
2. MacMahon H.E., Bedizel M., Ellis C.A. Familial erytrophagocytic limphohistiocytosis. Pediatrics 1963; 32: 868.
3. Bell R.I.M., Bratied A.I.E., Barnes N.D. Familial haemophagocytic reticulosis. Arch. Dis. Childh 1968; 43: 601.
4. Caiz J., Yancock B.W., Hanry J., Ward A.M. [Limforetikularnye bolezni, rus]. Moscow: Medicina; 1980.
5. Blennow Y., Berg B., Brandit L., at al. Haemophagocytic reticulosis: Astate chimirism? Arch. Dis. Childh 1979; 2: 1178.
6. Nezelof C., Eliachar E. La Limphocytose familiale Novcv. Rev. Franc. Hemat. 1973; 13: 319-338.
7. Gadnera H., Rumanceva A.G., editors. [Gistiocitozy detskogo vozrasta, rus]. Moscow-Vena: MaksPress; 2005.
8. Novikova G.A. [Gemofagocitarnye limfogistiocitozy u detej, rus] [dissertation]. Moscow; 1999.
9. Milovanova N.V. [Molekularno-geneticeskaa priroda pervicnyh gemofagocitarnyh limfogistiocitozov v Rossii, rus] [dissertation].Moscow; 2011.