
Международный неврологический журнал 5 (67) 2014
Вернуться к номеру
Спадкова мозочкова атаксія: особливості діагностики та перебігу
Авторы: Шульга О.Д. - Волинська обласна клінічна лікарня, м. Луцьк; Дешевцова О.М. - Окачинська центральна районна лікарня, Волинська область; Мельничук Т.В. - Волинська обласна клінічна лікарня, м. Луцьк
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
Розглядається клінічний випадок спадкової мозочкової атаксії, труднощі при діагностиці даного захворювання.
Рассматривается клинический случай наследственной мозжечковой атаксии, трудности в диагностике заболевания.
We consider the clinical case of hereditary cerebellar ataxia, difficulties in diagnosing this disease.
спадкова мозочкова атаксія.
наследственная мозжечковая атаксия.
hereditary cerebellar ataxia.
Статья опубликована на с. 139-142
Спадкові мозочкові атаксії (СМА) являють собою гетерогенну групу хвороб, що проявляються порушенням координації рухів і хиткістю при ході, часто порушенням вимови [3, 5]. Атаксія виникає внаслідок порушення функції мозочка та його зв’язків, ураження спинного мозку, периферичних сенсорних розладів та за рахунок комбінації цих причин. У більшості досліджених популяцій поширеність захворювання становить у середньому 1 на 100 тис. населення. У той же час у різних географічних зонах існують різні регіони, де частота захворювання зростає [4]. Група автосомно-домінантних спіноцеребелярних атаксій включає ряд самостійних захворювань, що є подібними за своєю клінічною картиною, основними патоморфологічними змінами і характером генетичного дефекту.
Клінічна картина СМА характеризується розвитком мозочково-пірамідного синдрому в ранньому дорослому віці [1, 5], його швидким прогресуванням, що призводить до соціальної дезадаптації хворих і помітного зменшення тривалості життя. Кожна із форм домінантної спинномозочкової атаксії має певні особливості неврологічного синдрому й перебігу хвороби.
Основні форми СМА
СМА типу 1 починається зазвичай на 3–4-му десятилітті життя. Основним проявом СМА типу 1 вже на ранніх стадіях захворювання є поєднання мозочкових порушень, дизартрії й пірамідної симптоматики. Через деякий час хворі перестають ходити і втрачають здатність до самообслуговування.
СМА типу 2: неврологічна картина схожа до такої СМА типу 1, але важливо відмітити, що у хворих із СМА типу 2 набагато частіше відмічається згасання сухожильних рефлексів та статокінетичний тремор рук. Як і при СМА типу 1, у сім’ях із СМА типу 2 нерідко спостерігається феномен антиципації, особливо при передачі захворювання по чоловічій лінії.
СМА типу 3, або хвороба Мачадо — Джозеф, характеризується вираженим поліморфізмом клінічних проявів. Разом із мозочковою атаксією у хворих можуть спостерігатися екстрапірамідна симптоматика (дистонія й синдром паркінсонізму), аміотрофії, симптоми ураження пірамідного тракту. Важливим діагностичним фактором є зовнішня офтальмоплегія, фасцикуляції періоральної мускулатури та феномен випуклих очей.
СМА типу 4 — рідкісна форма автосомно-домінантних атаксій, для якої характерне поєднання прогресуючих координаторних порушень та сенсорної невропатії.
СМА типу 5, 6: розвиток захворювання зазвичай розпочинається в пізньому віці (після 45–55 років). У неврологічному статусі відмічається розвиток відносно ізольованої атаксії ходи. Темп прогресування СМА типів 5 і 6 повільний, хворі протягом десятиліть зберігають здатність до самообслуговування. Хвороба практично не впливає на тривалість життя.
СМА типу 7: специфічною ознакою є поєднання мозочково–пірамідного синдрому і прогресуючої дегенерації сітківки (аж до повної сліпоти). Важливо відмітити, що порушення зору у хворих може на кілька років передувати координаторним розладам. Форми СМА типів 8–13 описані лише в одиничних сім’ях, їх клінічна картина вивчена недостатньо [2].
Диференціальна діагностика спадкових мозочкових атаксій проводиться з атаксією спадкового характеру при алкоголізмі, авітамінозі, з розсіяним склерозом, первинними або метастазуючими пухлинами, паранеопластичним синдромом при раку яєчника та молочної залози, аномалією Арнольда — Кіарі, нормотензивною гідроцефалією, недостатністю кровообігу у вертебробазилярному басейні.
Діагностика
Важливе місце в діагностиці автосомно–домінантних спіноцеребелярних атаксій займають методи нейровізуалізації — КТ, МРТ (наявність атрофічного процесу і відсутність вогнищевих змін у речовині мозку). Одним із факторів, що підтверджує діагноз, є наявність сімейного анамнезу та невпинно прогресуючий характер захворювання.
Лікування
На сьогодні ефективне специфічне лікування автосомно-домінантних спіноцеребелярних атаксій відсутнє. Лікування здебільшого скероване на вирішення проблем із координацією рухів за допомогою реабілітаційних та фізіотерапевтичних методів, лікувальної фізкультури. При порушенні ходи використовують палиці, інвалідні коляски, за допомогою спеціальних приладів полегшують письмо, користування столовими приборами, при дизартрії користуються допомогою логопеда.
Наводимо власний клінічний випадок спадкової мозочкової атаксії.
Хворий В., 43 років, звернувся до невропатолога Локачинської ЦРЛ зі скаргами на виражене похитування при ході, порушення мови, загальну слабкість.
Анамнез захворювання. Вважає себе хворим більше ніж 10 років, відколи з’явилося похитування при ході, що поступово наростало. Близько 5 років тому відмітив погіршення мови, що повільно прогресувало. За останні 10 років відмічає поступове «схуднення» м’язів кінцівок та тулуба. За медичною допомогою не звертався.
Неврологічний статус. У свідомості. Орієнтований у часі, просторі, власній особистості, контактний, адекватний. Очні щілини, зіниці D = S, фотореакції живі. Слабкість конвергенції з обох сторін. Горизонтальний ністагм при погляді в сторони. Злегка згладжена права носогубна складка. Язик по середній лінії. Акт ковтання не порушений. Виражена дизартрія. Сухожилкові рефлекси живі, симетричні. Патологічні стопні знаки відсутні. Сила та тонус збережені. Дифузна гіпотрофія м’язів кінцівок і тулуба. Чутливі розлади відсутні. Координаторні проби виконує з вираженою інтенцією. Виражене похитування в позі Ромберга. Хода атактична. Менінгеальні симптоми відсутні. Функцію тазових органів контролює.
Результати додаткових методів обстеження
Загальний аналіз крові (04.04.13 р.): Hb — 154 г/л, Er — 4,78 • 1012/л, КП — 1,0, Le — 9,3 • 109/л, ШОЕ — 2 мм/год, п — 3, с — 80, л — 17. Глюкоза крові (04.04.13 р.) — 4,7 ммоль/л. Загальний аналіз сечі (04.04.13 р.) — солом’яно–жовта, білка й цукру не виявлено, Le — 2–4 у полі зору. Я/г не виявлено (04.04.13 р.). RW негативна (04.04.13 р.).
ЕКГ (01.04.13 р.) — без особливостей.
ФОГК (01.04.13 р.) — без особливостей.
Консультація окуліста (01.04.13 р.). Очне дно: ДЗН бідо-рожеві, з чіткими контурами, калібр і хід судин не змінені. Гострота зору обох очей — 1,0.
Мати хворого, хвора В., 67 років, лікувалась у неврологічному відділенні Локачинської ЦРЛ. З анамнезу відомо, що близько 9 років тому з’явилося виражене похитування при ході, порушилась вимова. Останні 6 місяців мали місце часті падіння при ході. Протягом останніх 3 місяців майже не ходить, нерозбірливо розмовляє. При надходженні скарги на виражене утруднення вставання з ліжка та ходи (потребує підтримки сторонніх осіб, встає з опорою на руки), порушення мови, загальну слабкість, швидку втому.
За медичною допомогою не зверталася багато років. Згідно із записами в амбулаторній картці близько 10 років тому було діагностовано гіпертонічну хворобу. 9 років тому перенесла закриту черепно-мозкову травму (струс головного мозку). 8 років тому лікувалася в неврологічному відділенні з приводу дисциркуляторної енцефалопатії ІІ ст. із статико-координаторними розладами на ґрунті гіпертонічної хвороби ІІ ст.
Неврологічний статус. У свідомості. Орієнтована в часі, просторі, власній особистості, контактна, адекватна. Очні щілини, зіниці D = S, фотореакції живі. Парез погляду вгору. Слабкість конвергенції з обох сторін. Згладжена права носогубна складка. Легка девіація язика вправо. Ковтання не порушене. Виражена дизартрія. Сухожилкові рефлекси живі, симетричні. Патологічні стопні знаки відсутні. Сила та тонус м’язів кінцівок збережені. Чутливих розладів не виявлено. Координаторні проби виконує з вираженою інтенцією. Виражена статична атаксія (встає лише з опорою на руки, в позі Ромберга — виражене хитання, не може встояти без опори, хода атактична, з двома палицями). Менінгеальні симптоми відсутні. Функцію тазових органів контролює.
Результати додаткових методів обстеження
Загальний аналіз крові (27.03.13 р.): Hb — 142 г/л, Er — 4,23 • 1012/л, КП — 1,0, Le — 7,3 • 109/л, ШОЕ — 6 мм/год, п — 2, с — 66, е — 3, м — 4, л — 25. Загальний аналіз сечі (02.04.13 р.) — прозора, солом’яно-жовта, білка і цукру не виявлено, Le — 2–3 у полі зору, кристали сечової кислоти — у незначній кількості. Біохімічний аналіз крові (27.03.13 р.): загальний білок — 78,8, фібриноген — 4,66, холестерин — 5,17, сечовина — 8,5, креатинін — 100,7, білірубін загальний — 20,5, прямий — 7,3, непрямий — 13,2, АлАТ — 0,37, АсАТ — 0,5, СРБ (+). Глюкоза крові — 5,0 ммоль/л. Коагулограма: протромбіновий індекс — 79 %, протромбіновий час — 19′′, МНВ — 1,5. Я/г не виявлено (02.04.13 р.). RW негативна (29.03.13 р.).
ЕКГ (27.03.13 р.) — гіпертрофія лівого шлуночка, патологічні зміни в міокарді.
Рентгенографія ОГК (28.03.13 р.): легеневі поля емфізематозні, із посиленим судинним рисунком фіброзного характеру. Справа у проекції медіального діафрагмального синуса — обмежений кардіогенний плевропневмофіброз, а зліва у базальному відділі — ніжні плевропневмофіброзні тіньові нашарування. Корені малоструктурні, фіброзні.
Гінеколог (02.04.13 р.): здорова.
Консультація окуліста (29.03.13). Очне дно: ДЗН із чіткими контурами, дещо бліді, артерії звужені, вени розширені, повнокровні. Діагноз: ангіопатія сітківки обох очей.
Консультація терапевта (27.03.13 р.) — ІХС. Дифузний кардіосклероз. Застійна пневмонія. СН ІІА. Гіпертонічна хвороба ІІ ст., ст. 2, серцево-судинний ризик високий. Гіпертензивне серце. ПДОА гомілковостопних, колінних суглобів.
Спадковий анамнез. При зборі спадкового анамнезу було з’ясовано, що бабуся хворого, рідна тітка, дядько та двоюрідний брат по материнській лінії мали «хворобу ніг», у зв’язку з чим із часом перестали ходити. Двоюрідний брат захворів на «хворобу ніг» у юнацькому віці, помер до досягнення 30-річного віку. Скарги на порушення ходи наявні в рідної сестри пацієнта.
Наводимо схему родоводу даної родини (рис. 1). У даному родоводі має місце автосомно-домінантний тип успадкування.
/141/141.jpg)
Установлено попередній діагноз: спадкова мозочкова атаксія з вираженими статико-координаторними розладами та дизартрією. Хворий разом з матір’ю направлений для дообстеження у ВОКЛ.
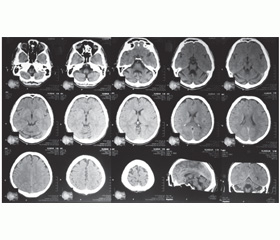
Проведено МСКТ голови (03.04.2013 р.) (рис. 2).
На серії томограм відзначається виражена картина атрофії гемісфер мозочка, помірна атрофія кори головного мозку на рівні лобних долей. Шлуночкова система не розширена. Зон патологічної щільності не відмічається.
Консультований неврологом ВОКЛ (03.04.2013). Установлено заключний діагноз: спадкова мозочкова атаксія з вираженими статико-координаторними порушеннями, дизартрією.
Консультований генетиком (03.04.2013). Діагноз: спадкова мозочкова атаксія, тип спадкування — автосомно-домінантний.
У зв’язку з відсутністю матеріальних та технічних можливостей генотипування не проводилося.
Висновок. Діагностика спадкових мозочкових атаксій вимагає детального збору спадкового анамнезу та проведення нейровізуалізації.
1. Иллариошкин С.Н. Пресимптомная ДНК-диагностика спиноцеребеллярной атаксии 1-го типа / С.Н. Иллариошкин, И.А. Иванова-Смоленская, С.А. Лимборская и др. // Генетика. — 1997. — Том 33, № 5. — С. 693–698.
2. Яхно Н.Н. Болезни нервной системы / Н.Н. Яхно, Д.Р. Штульман, П.В. Мельничук и др. // Медицина. — 2001. — Т. 2. — С. 164–171.
3. Pietrini V. Clinical and genetic study of a family with spinocerebellar ataxiatype 1 (SCA1) and beta-thalassemia / V. Pietrini, М. Godani, S. Calzetti // Ital. J. Neurol. Sci. — 1998 Dec. — 19(6). — 345–50.
4. Rich S.S. Spinocerebellar ataxia: localization of an autosomaldominant locus between two markers on human chromosome 6 / S.S. Rich, P. Wilkie, L. Schut. // Am. J. Hum. Genet. — 1987. — 41. — 524–8.
5. Storey E. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia / E. Storey, D. du Sart, J.H. Shaw et al. // Am. J. Med. Genet. — 2000 Dec 11. — 95(4). — 351–7.