Статья опубликована на с. 188-192
Описанный французским врачом D. Louis-Bar, а позднее — американскими врачами E. Boder, R.P. Sedgwick синдром (синоним — атаксия-телеангиэктазия) является наследственным, и начало его, как правило, отмечается с раннего детского возраста. У большинства детей наблюдается задержка психомоторного развития. Манифестация заболевания начинается с неврологической симптоматики: мозжечковой атаксии с нарушением походки, дрожания головы и туловища, хореоатетоза, интенционного тремора, нарушения движения глазных яблок. Примерно к 2–6 годам на коже тела и слизистых появляются асимметричные телеангиэктазии, нарушается пигментации кожи. Синдром Луи-Бар характеризуется врожденной ангиопатией с изначальным возникновением телеангиэктазий на конъюнктиве глазного яблока. Затем сосудистые «звездочки» появляются на коже век, носа, лица, шеи и конечностях.
Помимо сочетанного поражения кожи и нервной системы, синдром Луи-Бар характеризуется комбинированным иммунодефицитом, что обусловливает у больных склонность к частым инфекционным и онкологическим заболеваниям, формирование хронических бронхолегочных процессов. В иммунограмме максимально снижен или отсутствует сывороточный иммуноглобулин А, иногда G и E. Кроме того, у больных с синдромом Луи-Бар наблюдаются низкий уровень α-фетопротеина в крови, лимфопения и эозинофилия. Цитогенетическое исследование лимфоцитов показывает хромосомную нестабильность. Патоморфологически выявляют гипоплазию или аплазию вилочковой железы, лимфатических узлов, селезенки [1, 2].
В неврологии синдром Луи-Бар относится к так называемым факоматозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности прогрессирующей гибелью клеток Пуркинье. Дегенеративные изменения могут затрагивать и другие структуры головного мозга. При обследовании у пациентов с синдромом Луи-Бар на компьютерной и магнитно-резонансной томографии (МРТ) обычно определяется атрофия червя и полушарий мозжечка с расширением ликворных путей [3].
Синдром Луи-Бар наследуется по аутосомно-рецессивному типу, связан с мутациями в гене АТМ (ataxia telangiectasia mutated), основная функция которого заключается в репарации разрывов ДНК и поддержании целостности генома в клетке. Ген АТМ картирован на длинном плече хромосомы 11 в участке 11q22-q23 [4]. Предполагается, что гипоплазия тимуса и изменения нервной системы могут быть вызваны аномалиями сосудов, обусловленными мезодермальным дефектом, или аутоиммунными реакциями на антиген, общий для тимоцитов и нервных клеток. Однако причинная связь между иммунными, сосудистыми и неврологическими нарушениями не доказана.
Нестабильность генома на хромосомном уровне является генетической причиной не только синдрома Луи-Бар, но и других нарушений центральной нервной системы. Известно, что регулярные и мозаичные формы хромосомных болезней всегда негативно затрагивают функции головного мозга и проявляются в виде различных форм умственной отсталости, аутизма и эпилепсии [4, 5]. Однако в современной литературе последних лет встречаются лишь редкие описания тяжелых форм эпилепсии при атаксии-телеангиэктазии, в частности синдром Леннокса — Гасто, синдром Агилара [6, 7].
Сочетание эпилепсии с редкими наследственными или врожденными заболеваниями может вызывать существенные трудности в диагностике последних, значительно утяжеляя их прогноз. Это обусловливает необходимость индивидуального подхода к пациентам с неврологической патологией и проведения их комплексного обследования для выявления сопутствующих заболеваний.
Собственное наблюдение
В нашей клинической практике наблюдался случай сочетания синдрома Луи-Бар с эпилепсией у девочки-подростка. Пациентка длительное время наблюдалась в инфекционном отделении ДГКБ № 2, где она неоднократно находилась на лечении по поводу респираторного заболевания.
У пациентки К., 1999 года рождения, был верифицирован клинический диагноз «синдром Луи-Бар. Фокальная симптоматическая эпилепсия с частыми приступами в виде сложных абсансов и сложных парциальных приступов, умственная отсталость в стадии имбецильности. Хронический гнойный эндобронхит. Хронический пиелонефрит. Гипохромная анемия средней степени тяжести. Задержка физического развития».
Из анамнеза известно, что девочка родилась от 2-й беременности, 2-х родов. Данных о течении беременности нет. В родах оценка по шкале Апгар 7–8 баллов. Показатели массы и длины тела при рождении — средние (3400 г, 51 см). Со 2-го месяца жизни вскармливалась искусственно, коровьим молоком. Отставание в физическом и психомоторном развитии было отмечено уже на первом году жизни. Ребенок удерживал голову с 3 месяцев, поворачивался с живота на спину с 5 месяцев, сидел самостоятельно с 7 месяцев. Однако отмечалось более позднее развитие навыков ползания — с 1 года, ходьбы — с 2,5 года, с поддержкой, неуверенно, пошатываясь. Также отмечалась задержка развития речи — первые слова появились в 2-летнем возрасте. С рождения девочка неоднократно болела острыми респираторными вирусными инфекциями (ОРВИ), бронхитом, стрептодермией, энтероколитом. В 2-летнем возрасте перенесла сепсис, септикопиемическую форму, двустороннюю бронхопневмонию, двусторонний катаральный отит, гепатит. Ребенок рос в неблагополучной семье, где родители злоупотребляли алкоголем. Отставание в нервно-психическом и физическом развитии, а также частую инфекционную заболеваемость врачи связывали с дефектами ухода и вскармливания пациентки.
В 3-летнем возрасте девочке был выставлен диагноз «детский церебральный паралич, атактическая форма». Позднее появились такие симптомы, как пошатывание головы, туловища, сходящееся косоглазие, дизартрия, гипотония мышц конечностей. Педиатры и неврологи отмечали, что заболевание протекает с отрицательной динамикой: у девочки нарастали проявления мозжечковой атаксии с постепенной утратой навыков передвижения и самообслуживания, наблюдалась задержка психоречевого развития, дизартрия с уменьшением словарного запаса. Кроме того, тщательный анализ истории развития пациентки выявил факты периодического появления у нее высыпаний на лице и туловище, которые расценивались как проявление стрептодермии. Однако с 3-летнего возраста описание кожных элементов характеризовало их как «звездчатую» сыпь, что является характерным для синдрома Луи-Бар.
Только в 2010 году (в возрасте 11 лет), учитывая отрицательную динамику и атипичное течение основного заболевания, наличие множественных стигм дизэмбриогенеза, а также для уточнения диагноза девочка была направлена на консультацию в областной медико-генетический центр, где выставлен диагноз «синдром Луи-Бар? Лейкодистрофия?». Проведенное с целью дифференциальной диагностики МРТ головного мозга не выявило патологических изменений. Однако иммунологическое исследование установило лимфопению, значительное снижение IgA и IgЕ, что позволило подтвердить диагноз первичного иммунодефицита — «синдром Луи-Бар».
Особенностью данного клинического случая явилось сочетание синдрома Луи-Бар с эпилепсией и хроническим пиелонефритом. С июня 2010 г. клиническая картина заболевания усугубилась появлением судорожного синдрома. Неврологом диагностирована эпилепсия с частыми приступами в виде сложных абсансов и сложных парциальных приступов. В том же году на фоне острого вульвовагинита и дисбиоза кишечника были выявлены клинико-лабораторные симптомы пиелонефрита.
Пациентка К. с сентября 2009 г. проживает в Днепропетровском детском доме-интернате. В связи с рецидивирующими респираторными заболеваниями неоднократно поступала на лечение в инфекционное отделение ДГКБ № 2. За период наблюдения с 2009 года по настоящее время девочка неоднократно болела ОРВИ, бронхитами, трижды перенесла внегоспитальную бронхопневмонию. Особенностями течения респираторных заболеваний были затяжное течение, длительно сохраняющиеся аускультативные изменения в легких в виде локальных влажных, мелко- и среднепузырчатых хрипов, что позволило заподозрить хронизацию воспалительного процесса. Проведенное бронхологическое исследование подтвердило наличие морфологических признаков хронического диффузного гнойного эндобронхита.
Хронизация соматических заболеваний с формированием хронического бронхита и пиелонефрита на фоне врожденного иммунодефицитного состояния — синдрома Луи-Бар, частое рецидивирование обострений во многом определило и тяжесть состояния пациентки по неврологическому статусу. Наблюдалось учащение приступов эпилепсии и нарастание признаков деградации личности ребенка.
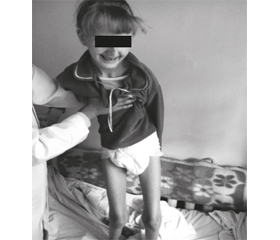
На сегодняшний день возраст пациентки 15 лет. При клиническом обследовании девочка эмоционально лабильна, заторможена, отвечает только на элементарные вопросы. Выражена дизартрия, голос тихий, словарный запас 10–12 слов, иногда отвечает жестами. Когнитивные функции снижены. Физическое развитие дисгармоничное, микросомия, резко выражена дистрофия. Мышечный тонус значительно снижен во всех группах, мышцы конечностей атрофированы. Чувствительность не нарушена. Садится самостоятельно. Стоит с поддержкой. Не ходит (рис. 1). Кушать может самостоятельно, но быстро устает. Может писать буквы, любит рисовать, однако наблюдается прогрессирующее ухудшение письма (рис. 2). Отмечаются сходящееся косоглазие, нистагм, сколиоз грудного и поясничного отделов позвоночника, нарушение функции тазовых органов в виде энуреза. Выражена гиперсаливация.
/190.jpg)
Кожные покровы бледные, сухие, наблюдаются множественные элементы «звездчатой» сыпи. На лице, шее — телеангиэктазии. На глазных яблоках выражен сосудистый рисунок, наблюдается инъекция склер. Периферические лимфатические узлы мелкие, единичные, до 0,8–1,0 см в диаметре. Со стороны дыхательной системы отмечались катаральные проявления в носоглотке, редкий, малопродуктивный кашель, одышка смешанного типа. Над легкими перкуторно определялся легочный звук, несколько укороченный в нижних отделах, аускультативно дыхание жесткое, в нижних отделах — ослабленное, выслушиваются проводные и единичные влажные, мелкопузырчатые хрипы слева в нижних отделах.
Данные дополнительного обследования: были выявлены гематологические изменения в виде гипохромной анемии І степени, лейкоцитоза, нейтрофилеза, повышения скорости оседания эритроцитов. В общем анализе мочи — лейкоцитурия и микропротеинурия. Рентгенологически подтверждены левосторонняя нижнедолевая пневмония, хронический бронхит.
Ультразвуковое исследование почек выявило эхо-признаки нефропатии: неоднородность чашечно-лоханочной системы с наличием мелких гиперэхогенных включений, некоторое расширение чашечки слева. Биохимическое исследование крови подтвердило нарушение азотовыделительной функции почек на основании высоких показателей креатинина.
По данным эхоэнцефалографии: умеренная пульсация латеральных эхо-комплексов без четких латеральных особенностей; с обеих сторон признаки умеренных гипертензионных нарушений, cлева признаки гидроцефальных нарушений; очаговых изменений не выявлено.
Проведенное иммунологическое исследование крови выявило относительную лимфопению на фоне лейкоцитоза, нарушение иммунорегуляторного индекса, низкую фагоцитарную активность и значительное снижение показателей IgА (табл. 1).
Проводимое лечение включало длительную антибактериальную терапию, назначение муколитиков, бронхолитических препаратов, ингаляционной терапии. Была проведена санационная бронхоскопия с лаважем бронхов и введением антисептиков. Комплексная терапия сопутствующих заболеваний включала применение противосудорожных, метаболических препаратов; витаминов В1, В6; сосудистых и ноотропных препаратов; массаж конечностей с мазью траумель.
На фоне проводимого лечения, помимо нивелирования симптоматики пневмонии, отмечено улучшение общего состояния пациентки, а именно: уменьшение частоты и тяжести эпиприступов, повышение двигательной активности и эмоционального тонуса девочки, улучшение практических навыков самообслуживания.
Таким образом, приведенный случай демонстрирует сложность ранней диагностики синдрома Луи-Бар у детей, так как возможное его сочетание с патологией нервной системы затрудняет постановку диагноза. Несмотря на специфичность клинической картины этой формы первичного иммунодефицита, разные варианты развития и течения заболевания с постепенным проявлением патогномоничной симптоматики могут скрывать синдром Луи-Бар под маской различных форм мозжечковой атаксии.
Для своевременной верификации диагноза необходим тщательный анализ клинико-анамнестических данных в сочетании с дифференциальным поиском причин рецидивирования инфекционных заболеваний у детей с неврологической патологией. Комплексный подход к обследованию пациентов с неврологической патологией должен включать не только динамическое наблюдение узких специалистов, но и проведение иммунологических, инструментальных исследований, а также ДНК-диагностики. В каждом конкретном случае терапия должна быть ориентирована не только на коррекцию неврологических симптомов, но и на профилактику повторных инфекционных заболеваний, что значительно улучшает прогноз в отношении продолжительности и качества жизни пациентов с сочетанной патологией.

/191.jpg)