Статтю опубліковано на с. 163-166
Синдром Пендреда (глухота вроджена нейросенсорна і зоб, дисгормональний зоб, Pendred Syndrome) — спадкова хвороба, що характеризується порушенням біосинтезу тиреоїдних гормонів, проявляєтaься вродженим гіпотиреозом, зобом і глухонімотою (МКХ-10: дисгормональний зоб (E07.1), номер із каталогу МІМ — 274600 Pendred Syndrome; PDS).
Успадковується за автосомно-рецесивним типом. Уперше описаний англійським лікарем Vaughan Pendred (1869–1946). Частота синдрому Пендреда становить від 1 до 10 % усіх випадків спадкової глухоти. Так, частота синдрому Пендреда на Британських островах становить 7,5 випадку на 100 000 новонароджених [3], у скандинавських країнах — близько 1 випадку на 100 000 новонароджених [4]. Опубліковано принаймні 300 випадків синдрому Пендреда [5]. Очевидно, якщо використовувати для діагностики класичні клінічні критерії синдрому Пендреда, значне число випадків залишиться невиявленим. Варіабельна експресивність синдрому Пендреда, особливо його тиреоїдних проявів, може штучно занижувати реальну частоту захворювання. Співвідношення статей становить 1 : 1.
У 75 % пацієнтів виявляється дефект гена PDS PDS (SLC26A4, solute carrier family 26, member 4), локалізованого на довгому плечі 7-ї хромосоми (7q22-q31) [6]. Нормальний генний продукт — ген, що кодує білок із 780 амінокислот (86 kDa). Ген SLC26A4 кодує трансмембранний транспортний білок пендрин — багатофункціональний аніонний обмінник, що активно експресується в тканинах щитоподібної залози, внутрішнього вуха й нирок. Вважається, що генетичний дефект може бути пов’язаний не тільки з йодуванням тирозину, але і з перетворенням моно- й дійодтирозину в йодтироніни. Наявність вродженої глухоти при синдромі Пендреда і її відсутність при звичайному дефекті органіфікації зумовлюються плейотропною дією гена, що контролює біосинтез тиреоїдних гормонів і розвиток внутрішнього вуха.
Описано 47 різноманітних мутацій гена SLC26A4, що є причиною синдрому Пендреда. Три мутації L236P, T416P та 1001+1 Г на А є мажорними. Проведено аналіз мутацій у гені PDS у 14 сім’ях із синдромом Пендреда з 7 країн Європи і виявлено 14 різних мутацій (10 місенс-мутацій, 1 мутація сплайсингу й 3 мікроделеції). Дві з цих мутацій виявилися частими: мутація L236P виявлена у 7 сім’ях, а мутація T416P — у 4 сім’ях [4].
Основні діагностичні критерії:
— вроджена непрогресуюча нейросенсорна глухота;
— еутиреоїдний зоб;
— мальформація Мондіні або розширення вестибулярного каналу.
Клінічна діагностика: зоб здебільшого дифузний, м’якої консистенції, але в деяких випадках спостерігаються вузли. Характерна еутиреоїдна струма, однак можуть бути ознаки гіпотиреозу, особливо після тиреоїдектомії. У такому випадку у хворих спостерігається затримка росту, надлишкова вага, хоча ожиріння буває рідко, блідість, анорексія, сухість шкіри, затримка статевого розвитку, брадикардія. Інколи в пацієнтів з еутиреоїдним зобом спостерігається надмірна відповідь на введення тиреотропного гормона, що свідчить про компенсований гіпотиреоїдизм [2]. У понад 75 % пацієнтів зоб виявляється при ультрасонографічному дослідженні [6]. Після тиреоїдектомії вузли часто рецидивують, якщо не буде призначена тиреоїдна терапія. У новонароджених пацієнтів із синдромом Пендреда щитоподібна залоза не збільшена. У 40 % випадків зоб розвивається в старшому дитячому та пубертатному віці, іноді в юнацькому, решта випадків припадають на дорослий вік. У рідкісних випадках можлива розумова відсталість. Вік, у якому спостерігається збільшення щитоподібної залози, варіює навіть у пацієнтів з однієї сім’ї, що ускладнює диференціальну діагностику синдрому Пендреда й ізольованої приглухуватості. Формування зоба відбувається через порушення органіфікації йоду в процесі біосинтезу тиреоїдних гормонів у щитоподібній залозі.
Макроскопічно щитоподібна залоза збільшена в розмірах, хоч і меншою мірою, ніж при дифузному зобі. Поверхня залози зазвичай горбиста. На розрізі в товщі гіперплазованої тканини залози визначаються вузли різного розміру, дрібнозернистої будови, блідо-жовтого кольору, зазвичай у тонкій капсулі. У великих вузлах можливі вогнищеві некротичні зміни, масивні крововиливи, формування кістозних порожнин.
Мікроскопічно гіперпластичний процес у щитоподібній залозі виражається в утворенні вогнищ проліферації тиреоїдного епітелію мікрофолікулярно-солідної будови. Іноді виявляються досить великі осередки папілярної гіперплазії, що мікроскопічно аналогічні таким при дифузному токсичному зобі. Форма фолікулів зазвичай неправильна, іноді сильно сплощена. Тиреоїдні клітини — низькі з дрібними ядрами, однак у місцях проліферації зустрічаються великі тироцити з різко збільшеними гіпо- або гіперхромними ядрами.
Ступінь вираженості втрати слуху може бути різним. Класично — це білатеральна виражена прелінгвальна нейросенсорна глухота. Причиною прогресивної втрати слуху є дисплазії завитка (cochlеa) та розширення вестибулярних каналів. Синдром Пендреда — єдине відоме генетичне захворювання, для якого характерний широкий вестибулярний канал (понад 1,5 мм). Вестибулярні порушення спостерігаються в 66 % пацієнтів із синдромом Пендреда, коливаючись у широких межах від незначного однобічного ураження до повного випадіння функції. Недорозвинені перетинки в дистальних кільцях завитка (деформація Мондіні) були звичною, але непостійною ознакою.
У літературі є повідомлення про незвичайний розподіл пігменту в сітківці у 3 сибсів [4]. Однак інші автори не згадують про подібні зміни.
Важливе діагностичне значення мають:
— крива поглинання радіоактивного йоду — швидкий високий підйом через 2 години й різке зниження в подальшому;
— проба з перхлоратом або тіоціанідом калію (при непорушеному процесі органіфікації йоду щитоподібною залозою він становить менше від 10 %, у пацієнтів із синдромом Пендреда — значно зростає, сягаючи 15–80 %). Перхлоратний тест є досить чутливим діагностичним тестом (хибнонегативні результати не перевищують 3 %);
— знижені концентрації тироксину (Т4), трийодтироніну (Т3), бутанол-екстрагуючого йоду, зв’язаного з білком йоду, і підвищена концентрація тиреотропного гормона (ТГ);
— томографія скроневих кісток (виявляють аномалії кісток лабіринту: дисплазія Мондіні або розширення вестибулярного водопроводу);
— аудіометрія;
— дослідження функцій вестибулярного апарату;
— ДНК-діагностика (виявлення мутації у гені SLC26A4 у локусі 7q22-q31) — позитивна в 75 % випадків.
Таким хворим необхідна консультація сурдолога для визначення ступеня порушення слуху. Виділяють такі ступені:
1) легкий (26–40 dB);
2) помірний (41–55 dB);
3) помірно тяжкий (56–70 dB);
4) тяжкий (71–90 dB);
5) глибокий (> 90 dB).
Для того щоб визначити відсоток втрати слуху, від чистого тону частотою 500, 1000, 2000, 3000 віднімається 25 dB. Результат множиться на 15, щоб отримати вухоспецифічний рівень. Пошкодження визначається урівноваженням значень вуха, що краще чує, з п’ятикратним множенням значень вуха, що гірше чує.
Визначається також частотність приглухуватості:
1) низькочастотна (< 500 Hz);
2) середньочастотна (501–2000 Hz);
3) високочастотна (> 2000 Hz).
Аудіометричні дослідження зазвичай показують наявність двосторонньої вродженої нейросенсорної глухоти зі значно варіюючим ступенем втрати слуху в діапазоні 40–100 dB, різкіше вираженим на високих частотах. Більше ніж у 50 % випадків дефект слуху різко виражений. Середній вік, у якому глухота може бути визначена, становить 2,2 року. У дитинстві втрата слуху повільно прогресує. Легке ураження слуху спостерігається рідко, іноді може відзначатися майже нормальний слух на одне вухо. Мова зазвичай розвивається погано.
З урахуванням важливості раннього виявлення та своєчасної корекції порушень слуху програма скринінгу слуху всіх новонароджених здійснюється в даний час більше ніж у 50 країнах. Слуховий скринінг новонароджених є складовою частиною програми раннього виявлення та корекції порушень слуху в дітей, спрямованої на поліпшення якості життя дітей із виявленими порушеннями за допомогою запобігання вторинним порушенням мовлення, затримці мовного розвитку, низькій академічній успішності та психоемоційним розладам.
Стандарти здійснення програми раннього виявлення та корекції порушень слуху в дітей сформульовані Об’єднаною комісією зі слуху дітей (США) у 2007 році і є загальноприйнятими в усьому світі. Це так званий принцип 1–3–6, відповідно до якого:
— скринінг слуху проводять усім новонародженим до досягнення ними віку 1 місяця, оптимально до моменту їх виписки з пологового відділення;
— аудіологічна діагностика (визначення ступеня й характеру порушення слуху) при позитивних результатах скринінгу, тобто виявлення порушень слуху, проводиться всім дітям до досягнення ними віку 3 місяців;
— реабілітацію порушень слуху (слухопротезування) всім дітям із виявленими необоротними порушеннями слуху починають не пізніше досягнення ними віку 6 місяців.
В Україні програми раннього виявлення та корекція порушень слуху в дітей на даний час не впроваджені [1]. Налагоджений тільки об’єктивний слуховий скринінг на основі використання найбільш надійного методу скринінгу — реєстрації коротколатентних слухових викликаних потенціалів. Цей метод скринінгу дозволяє виявити порушення слуху, зумовлені ураженням середнього, внутрішнього вуха, слухового нерва й стовбура головного мозку.
Ускладнення:
— повна втрата слуху (глухота);
— розумова відсталість;
— синдром здавлювання оточуючих органів при значному збільшенні щитоподібної залози внаслідок зоба;
— стійкий гіпотиреоз із довічною терапією тиреоїдними гормонами.
Для ілюстрації вищевикладеного наводимо витяг з історії захворювання хлопчика Р., 2,2 року, батьки якого звернулися за консультацією в обласну дитячу клінічну лікарню м. Чернівців.
З анамнезу життя й захворювання відомо, що дитина від третьої вагітності, що перебігала на тлі анемії. Пологи другі, в терміні гестації 39 тижнів. Маса тіла при народженні 3500 грамів, на штучному вигодовуванні з народження. Хлопчик до року спостерігався в невролога з діагнозом гіпоксично-ішемічної енцефалопатії. У віці 1 року 4 місяців дитині оформлена інвалідність з приводу уродженої нейросенсорної глухоти. Серед перенесених захворювань — повторні гострі респіраторні вірусні інфекції, обструктивні бронхіти.
При огляді встановлено, що хлопчик відстає в психомоторному розвитку (стоїть і ходить із підтримкою, вимовляє окремі склади, намагаючись поєднати їх у слова, розуміє звернену до нього мову). Привертає увагу фенотип дитини: макроглосія, виступаючий лоб, широке перенісся, коротка шия. Маса тіла 15 кг. Зріст 80 см. Підшкірно-жирова клітковина розвинена надмірно, розподілена нерівномірно, з переважанням на грудях, животі, стегнах. Набряків немає. Тонус м’язів і тургор тканин задовільний. Шкірні покриви бліді, сухі, вільні від висипань. Періорбітальні тіні. Слизова зіва рожева, волога, чиста. Пальпуються підщелепні лімфатичні вузли — до 0,7–0,9 см у діаметрі, рухомі, безболісні. Носове дихання утруднене, виділень немає. Дихання 25 рухів за хвилину. Перкуторно над легенями легеневий звук, аускультативно — ослаблене дихання. Ділянка серця без видимих деформацій. ЧСС 100 поштовхів за хвилину. Тони серця ритмічні, помірно приглушені. Язик збільшений, дещо виступає із порожнини рота, чистий. Живіт м’який, доступний глибокій пальпації. Печінка +2,5 см нижче від краю реберної дуги, край еластичний, безболісний. Селезінка не пальпується. Випорожнення оформлені, без патологічних домішок. Сечовипускання вільне, безболісне. Діурез адекватний.
У клінічному аналізі крові: відносна лімфопенія (38 %), анемія (гемоглобін 92 Г/л, еритроцити 3,2 Г/л, КП 0,87). Клінічний аналіз сечі — в нормі. Біохімічне дослідження крові: креатинін крові 54,2 (N 27–62 мкмоль/л), сечовина крові 4,33 (N 1,8–6,4 ммоль/л); фосфор 1,50 (N 1,45–1,78 ммоль/л); магній 0,7 (N 0,7–0,95ммоль/л); кальцій 2,1 (N 2,02–2,6 ммоль/л); глюкоза крові 5,3 ммоль/л. Печінкові проби: АЛТ 22,7 МО/л (N 5–30 МО/л), АСТ 20,6 МО/л (N 7–40 МО/л), загальний білірубін 10,8 мкмоль/л (N 8,6–20,5 мкмоль/л), зв’язаний 3,7 мкмоль/л (N до 5,1 мкмоль/л), вільний 7,1 мкмоль/л (N до 15,4 мкмоль/л).
Аналіз крові на гормони: Т4 вільний 8,6 пмоль/л (N 9–25 пмоль/л), Т3 вільний 2,0 пмоль/мл (N 2,6–5,7 пмоль/л), тиреотропний гормон 10,03 мкМО/мл (N 0,23–3,4 мкМО/мл), ТЗГ 28,8т мг/л (N 25–50 мг/л), АТ ТГ 8 ОД/мл (N 0–18 ОД/мл), АТ ТПО 12 (N до 30 МО/мл).
ІФА на сумарні антитіла до гельмінтів: лямблії, токсокари, аскариди (АТ IgG негативний). Проба з перхлоратом калію позитивна — 65 % (N до 10 %).
Ультразвукове дослідження щитоподібної залози: об’єм 3,2 см3 (N 2,2 см3), ехоструктура не змінена, об’ємні утворення відсутні.
Ультразвукове дослідження органів черевної порожнини: печінка збільшена (вертикальний косий розмір 96 мм (N до 85 мм)), край гострий, контури гострі, структура однорідна, ехогенність не змінена. Жовчний міхур: 46 × 16 мм, перегин дна та шийки, стінки не потовщені, каменів немає. Підшлункова залоза — без особливостей. Селезінка: 71 × 32 мм, не збільшена. Нирки — права 61 × 26 мм, ліва 63 × 35 мм, паренхіма 13 мм, однорідна, чашково-мискова система не розширена, каменів немає. Сечовий міхур у межах норми.
ЕКГ: ЧСС = 102 скорочення за хвилину, синусова брадикардія, амплітуда зубців R, P та комплексу QRS знижена, негативний зубець Т.
Неповна блокада правої ніжки пучка Гіса. На ЕЕГ визначаються патологічні уповільнені хвилі та недостатня вираженість альфа-ритму. МРТ скроневих кісток — аномалія Мондіні.
Генетичне дослідження. Мутація q.919-2A>G (7-й інтрон у сайті сплайсингу 8-го екзону в гені SLC26A4.
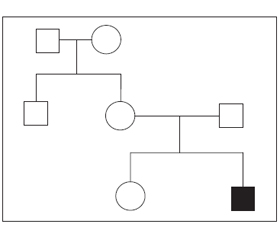
Невролог: затримка психомоторного розвитку. Ендокринолог: дифузний зоб. Гіпотиреоз. Офтальмолог: патології не виявлено. Сурдолог: двостороння вроджена нейросенсорна глухота тяжкого ступеня (80 dB), високочастотна (2020 Hz). Генетик: синдром Пендреда. На рис. 1 подано родовід пробанда.
Клінічний діагноз: синдром Пендреда. Гіпотиреоз.
Супутній: дефіцитна анемія II ступеня. Аномалія жовчного міхура.
Дитині призначено лікування: левотироксин 5 мкг/кг/добу, антианемічні засоби (препарати заліза, вітаміни В1 і В12, фолієва кислота); ноотропіл, ессенціалє, масаж, лікувальна фізкультура. Повторна аудіометрія з вирішенням питання про імплантацію завитка. Рекомендовано спостереження педіатра, ендокринолога, сурдолога, психоневролога двічі на рік. Медико-генетичне консультування пацієнта з синдромом Пендреда та його найближчих родичів, що може виявити хворих із синдромом Пендреда серед сибсів. Оскільки повторний ризик захворювання в сім`ї з ураженим пробандом є високим (25 %), рекомендується проведення пренатальної ДНК-діагностики шляхом біопсії хоріона на 10–12-му тижні вагітності. Важливим є запобігання виникненню ускладнень у хворих із синдромом Пендреда. З метою зменшення прогресуючої втрати слуху необхідно:
— уникати участі в контактних видах спорту;
— захищати голову від різних пошкоджень під час видів діяльності, при яких існує небезпека пошкодити голову;
— уникати ситуацій, при яких можливо отримати баротравму (різка зміна тиску), акустичну травму.
Висновок
Характеристика синдрому Пендреда включає: 1) автосомно-рецесивне успадкування; 2) зоб, що розвивається до пубертатного віку; 3) позитивну перхлоратну пробу вивільнення йоду; 4) симетричну, зазвичай глибоку вроджену нейросенсорну глухоту.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів.

/164.jpg)
/165.jpg)