Резюме
Незважаючи на успіхи в терапії цукрового діабету, частота виникнення ускладнень, зокрема порушення водно-електролітного та кислотно-основного стану, останніми роками не зменшилась. Цікаво відмітити той факт, що клінічні прояви такого грізного ускладнення, як діабетичний кетоацидоз, уперше описав Дрешфельд у 1886 році. Відтоді і до початку застосування інсуліну (в 1922 р.) смертність від цього ускладнення сягала 100 %. Широке впровадження в практику інсулінотерапії знизило смертність до 30 %, а з удосконаленням методів лікування, зокрема інфузійної терапії, відбулось подальше її зниження. На сьогодні рівень летальності пацієнтів із порушеннями водно-електролітного та кислотно-основного стану залишається високим (близько 5 % у спеціалізованих центрах). Тільки в США щорічно від цього ускладнення помирає близько 4000 хворих на цукровий діабет. Прогноз перебігу захворювання значно погіршується з віком, при розвитку коми та артеріальної гіпотензії. Поширеність цукрового діабету стрімко зростає і на сьогодні становить від 20 до 50 % випадків вперше виявленого захворювання в молодих людей. Порушення електролітів є частими в пацієнтів із даною патологією і можуть бути результатом зміненого розподілу електролітів, пов’язаного з індукованою гіперглікемією осмотичних зрушень рідини, або загального дефіциту, спричиненого осмотичним діурезом. Ускладнення від пошкодження органів-мішеней і методи лікування цукрового діабету можуть також сприяти електролітним порушенням. Порушення регуляції гомеостазу глюкози призводить до великої кількості прямих і непрямих впливів на електролітний і кислотно-основний баланс, оскільки велика поширеність діабету гарантує, що лікарі практично кожної спеціальності мають справу з пацієнтами, хворими на цукровий діабет, а знання, пов’язані зі змінами електролітів, є необхідними. У статті розглянуто, як впливають зміни гомеостазу глюкози при цукровому діабеті на водно-електролітний баланс та кислотно-основний стан пацієнта.
Несмотря на успехи в терапии сахарного диабета, частота возникновения осложнений, в частности нарушения водно-электролитного и кислотно-основного состояния, в последние годы не уменьшилась. Интересно отметить тот факт, что клинические проявления такого грозного осложнения, как диабетический кетоацидоз, впервые описал Дрешфельд в 1886 году. С тех пор и до начала применения инсулина (в 1922 г.) смертность от этого осложнения достигала 100 %. Широкое внедрение в практику инсулинотерапии снизило смертность до 30 %, а с совершенствованием методов лечения, в частности инфузионной терапии, произошло дальнейшее ее снижение. На сегодняшний день уровень летальности пациентов с нарушениями водно-электролитного и кислотно-основного состояния остается высоким (около 5 % в специализированных центрах). Только в США ежегодно от этого осложнения умирает около 4000 больных сахарным диабетом. Прогноз течения заболевания значительно ухудшается с возрастом, при развитии комы и артериальной гипотензии. Распространенность сахарного диабета стремительно растет и в настоящее время составляет от 20 до 50 % случаев впервые выявленного заболевания у молодых людей. Нарушения электролитов являются частыми у пациентов с данной патологией и могут быть результатом измененного распределения электролитов, связанного с индуцированной гипергликемией осмотических сдвигов жидкости, или всеобщего дефицита, вызванного осмотическим диурезом. Осложнения от повреждения органов-мишеней и методы лечения сахарного диабета могут также способствовать электролитным нарушениям. Нарушение регуляции гомеостаза глюкозы приводит к большому количеству прямых и косвенных воздействий на электролитный и кислотно-основной баланс, поскольку большая распространенность диабета гарантирует, что врачи практически каждой специальности имеют дело с пациентами, больными сахарным диабетом, а знания, связанные с изменениями электролитов, являются необходимыми. В статье рассмотрено, как влияют изменения гомеостаза глюкозы при сахарном диабете на водно-электролитный баланс и кислотно-основное состояние пациента.
Despite the success of the treatment of diabetes mellitus, the incidence of diabetic ketoacidosis in recent years has not decreased. Its clinical manifestations Dreshfeld first described in 1886. Insulin began to be used in 1922. Mortality of this complication was 100 %. Widespread implementation of insulin therapy into the clinical practice reduced mortality by 30 %, and with improvement of treatment, including infusion therapy, there was a further reduction. The level of mortality of patients with diabetic ketoacidosis remains high (about 5 % in specialized centers). Prediction of the disease much worsens with age, in the development of coma and hypotension. The prevalence of diabetes mellitus is growing rapidly and currently ranges from 20 to 50 % of new-onset disease in young people. Violations of electrolytes are frequent in patients with this disease and may be the result of altered distribution of electrolytes associated with hyperglycemia-induced osmotic fluid shifts or general shortages caused by osmotic diuresis. Complications from damage to the target organs and therapies used in the treatment of diabetes can also contribute to electrolyte disturbances. We highlight the ways, in which specific electrolytes may be influenced by dysregulation in glucose homeostasis. High or normal plasma sodium concentration in the presence of hyperglycemia indicates a clinically significant deficit of the total body fluid. Sodium correction in patients with glycemia helps to evaluate shortage of sodium and water, and ensure adequate assessment required for tonicity during the course of rehydration therapy. Because children with diabetic ketoacidosis are at particularly high risk of such terrible complications, as swelling of the brain, then to some extent hypernatremia is acceptable during treatment to minimize this complication. Insulin deficiency, which often occurs in type 1 diabetes mellitus, is an important factor of potassium output from the cells. Changes in potassium level, caused by metabolic acidosis, are more significant in mineral acidosis (hyperchloremic, non-anion gap acidosis) than in the organic acidosis (large anion gap acidosis), which is present in diabetic ketoacidosis. In diabetic patients receiving non-selective β-blockers, increased adrenergic activity can exacerbate hyperkalemia, because unopposed α-receptor stimulation, promotes the release of potassium from the cells. Ketoacidosis occurs when the rate, at which hepatic ketoacid is generated, exceeds peripheral utilization and the concentration of ketoacid in the blood increases. Normalization of acid-base balance is made within a few days, as the correction of bicarbonate deficit occurs through its regeneration by kidneys. Patients with primary kidney diabetic ketoacidosis are not involved (with their normal function). Kidney compensate by higher ammonia discharge. Urinary osmolarity gap is measured in order to find out whether there is a corresponding increase in the excretion of ammonia, which leads to violations in acid-base status. Low urinary osmolarity gap in patients with persistent hyperchloremic acidosis provides leads to tubular dysfunction. Despite recent advances that allowed direct assessment of the levels of β-hydroxybutyrate, determining the concentration of ketone bodies often can not fully explain the increase in anion gap and in many patients may contribute to hyperlactatemia. Lactate level may be increased in response to increased adrenergic activity, even in the absence of tissue hypoperfusion. Chronic hyperglycemia leads to the development of «carbonyl stress» and accumulation of toxic dicarbonyl compounds, such as methyl glyoxal, etc. Epidemiological studies suggest that low magnesium intake is associated with increased risk of diabetes, while higher magnesium intake is associated with lower risk of developing diabetes. In patients with diabetic ketoacidosis, osmotic diuresis due to poor glycemic control leads to renal loss of magnesium. Hypomagnesemia can cause hypocalcemia, because magnesium deficiency may lead to lower parathyroid hormone release. Randomized studies of phosphate therapy in patients with diabetic ketoacidosis did not establish that this therapy provides clinical benefit. The dysregulation of glucose homeostasis leads to many direct and indirect effects on electrolyte and acid-base balance. Since the high prevalence of diabetes ensures that clinicians of every medical specialty will interact with these patients, familiarity with related electrolyte abnormalities is important.
Статтю опубліковано на с. 16-26
Робота є фрагментом кафедральної планової науково-дослідної роботи «Клініко-патофізіологічні аспекти анестезіологічного забезпечення оперативних втручань та інтенсивної терапії у хворих із порушенням гомеостазу».
Незважаючи на успіхи в терапії цукрового діабету (ЦД), частота виникнення ускладнень, –зокрема порушення водно-електролітного та кислотно-основного стану, останніми роками не зменшилась. Цікаво відмітити той факт, що клінічні прояви такого грізного ускладнення, як діабетичний кетоацидоз (ДКА), уперше описав Дрешфельд у 1886 році. Відтоді і аж до початку застосування інсуліну (в 1922 р.) смертність від цього ускладнення сягала 100 %. Широке впровадження в практику інсулінотерапії знизило смертність до 30 %, а з удосконаленням методів лікування, зокрема інфузійної терапії, відбулось подальше її зниження [51]. На сьогодні рівень летальності пацієнтів із ДКА залишається високим (близько 5 % у спеціалізованих центрах). Тільки в США щорічно від цього ускладнення помирає близько 4000 хворих на ЦД [30]. Прогноз перебігу захворювання значно погіршується з віком, при розвитку коми та артеріальної гіпотензії.
Поширеність ЦД стрімко зростає і на сьогодні становить від 20 до 50 % випадків вперше виявленого захворювання в молодих людей [36]. Порушення електролітів є частими в пацієнтів із даною патологією і можуть бути результатом зміненого розподілу електролітів, пов’язаного з індукованою гіперглікемією осмотичних зрушень рідини, або загального дефіциту, спричиненого осмотичним діурезом. Ускладнення від пошкодження органів-мішеней і методи лікування, що використовуються в лікуванні ЦД, можуть також сприяти електролітним порушенням. Розглянемо, як впливають зміни гомеостазу глюкози при ЦД на водно-електролітний баланс та кислотно-основний стан пацієнта.
Натрій. Підвищення концентрації глюкози в плазмі крові може призвести до змін концентрації натрію через кілька механізмів. Зріст концентрації глюкози збільшує тонічність плазми, роблячи осмотичний тиск рушійною силою води з внутрішньоклітинного простору в позаклітинний, що спричинює розведення позаклітинної концентрації натрію. Концентрація натрію в плазмі зазвичай низька за рахунок осмотичного потоку води, а висока чи нормальна його концентрація за наявності гіперглікемії вказує на клінічно значимий дефіцит загальної рідини організму. Практичні рекомендації щодо ведення гіперглікемічних криз у дорослих пропонують додавати поправний коефіцієнт 1,6 мг/дл до визначеної концентрації натрію плазми на кожні 100 мг/дл (5,6 ммоль/л) глюкози, вище 100 мг/дл — для обліку розведеного ефекту глюкози [16, 30]. Корекція натрію в пацієнтів із глікемією допомагає оцінити дефіцит натрію та води і забезпечує адекватну оцінку необхідної тонічності протягом курсу регідратаційної терапії. Поправні коефіцієнти варіюють від 1,35 до 4,0 ммоль/л [22, 51]. Така мінливість у діапазоні корекції натрію виникає, швидше, через те, що в пацієнтів зі збереженою нирковою функцією є відкрита гіперглікемічна система, яка вводить ряд змінних, які тяжко визначити кількісно, а тому робить застосування стандартної корекції неточним фактором. Слід наголосити на тому, що скоригована концентрація натрію під час вибірки не враховує ефектів осмотичного діурезу та прийому рідини під час лікування, обидва з яких є значно різноманітними та непередбачуваними. Часті розрахунки скоригованої концентрації натрію разом із ретельним контролем діурезу є необхідними під час лікування всіх гіперглікемічних кризів. Стабілізація гемодинаміки фізіологічним розчином є первинною метою інфузійної терапії в таких пацієнтів. Протягом лікування можливий перехід до більш гіпотонічних розчинів, коли визначено дефіцит загальної води організму. При скоригованій концентрації натрію в плазмі вливання 0,9% фізіологічного розчину повинно бути продовжене [16, 30, 38]. Як нещодавно заявили Kamel та Halperin, розчини для регідратації повинні звести до мінімуму зменшення ефективної осмолярності, зокрема, протягом перших 15 годин терапії, для того щоб знизити ризик набряку мозку [27]. Через те, що діти з ДКА мають особливо високий ризик розвитку такого грізного ускладнення, як набряк головного мозку, то певною мірою гіпернатріємія прийнятна під час курсу лікування, щоб мінізувати це ускладнення.
Деякі препарати, що застосовуються при лікуванні ЦД, також асоціюються з гіпонатріємією [41]. Трициклічні антидепресанти, що використовуються при лікуванні діабетичної нейропатії, як відомо, стимулюють вивільнення вазопресину. –Пероральні гіпоглікемічні препарати, такі як хлорпропамід і толбутамід, можуть призвести до гіпонатріємії шляхом збільшення ефекту ендогенного вазопресину на рівні збірних канальців (трубок) нирок. Інсулін стимулює аргінін-вазопресин-залежну експресію аквапуринів-2 у збірних ниркових канальцях та збільшення гідроосмотичного ефекту вазопресину, коли ці циркулюючі рівні збільшуються у відповідь на інший вплив [7]. Останній ефект пояснює виявлений зв’язок між використанням інсуліну та нозокоміальною гіпонатріємією в пацієнтів із ЦД [5]. Гіпонатріємія також може розвинутися, якщо пацієнт із нелікованим діабетом мав гіпертригліцеридемію, навіть за наявності нормального рівня натрію, — це явище називають псевдогіпонатріємією [32].
Калій. Дефіцит інсуліну, що частіше спостерігається при ЦД 1-го типу, є важливим фактором при виході калію з клітини. У хворих на ЦД 2-го типу інсулінопосередковане поглинання глюкози порушується, але клітинне поглинання калію залишається нормальним шляхом активації інсулінових рецепторів, а тому узгоджується з дивергенцією внутрішньоклітинних шляхів [37]. Гіперкаліємія може бути спричинена збільшенням тонічності (осмолярності) плазми, що є результатом перерозподілу калію з внутрішньоклітинного простору в позаклітинний [42]. Вихід калію з клітини відбувається через внутрішньоклітинну дегідратацію, що є наслідком осмотично індукованого трансцелюлярного руху води, і власне він створює сприятливий градієнт для відтоку калію [58]. Введення розчину глюкози як короткострокової терапії при гіперкаліємії без супутнього введення інсуліну може погіршити гіперкаліємію в пацієнтів із ЦД, оскільки ендогенна секреція інсуліну може бути недостатньою чи непередбачуваною і тим самим призвести до збільшення тонічності плазми.
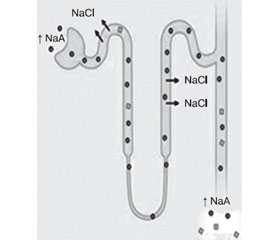
Гіперкаліємія часто наявна при надходженні пацієнтів із ДКА у відділення інтенсивної терапії, при тому що загальний рівень калію знижується. Цей стан викликаний виснаженням калію від збільшення доставки натрію щодо дистального нефрона в поєднанні з підвищеною мінералокортикоїдною активністю [43, 46] (рис. 1).
/16-26/18-1.jpg)
У регуляції об’єму артеріальної крові (блок А) існує збалансований, обернений зв’язок між постачанням натрію стосовно дистального нефрона та циркуляцією альдостерону, що необхідний для підтримки балансу калію. У пацієнтів із нелікованим діабетом (блок Б) осмотична діуретична дія глюкози (глюкоза Тmax — це максимальна швидкість реабсорбції глюкози в проксимальних канальцях) і екскреція солей натрію кетокислот стимулюють збільшення доставки натрію до дистального нефрона, тоді ж мінералокортикоїдна активність підвищується у відповідь на зниження об’єму. Поєднання збільшення доставки натрію з високою мінералокортикоїдною активністю в нирках призводить до виснаження калію та організму взагалі. Використання петльових чи тіазидних діуретиків також призводить до ниркового зниження калію. Високий потік у дистальному нефроні також активує секрецію калію за допомогою кальцій-активованого калієвого каналу (або максі-K+-канал).
У цих умовах гіперкаліємія спричинена перерозподілом калію, що є результатом гіпертонічності й інсулінового дефіциту, а не метаболічного ацидозу. Зміни калію, спричинені метаболічним ацидозом, більш яскраві при мінеральному ацидозі (гіперхлоремічний ацидоз без аніон-розриву), ніж при органічному (ацидоз із великим аніон-розривом), що є при ДКА [3]. Крім того, зміни калію, що є результатом гіпертонічності та дефіциту інсуліну, врівноважуються помітною активацією симпатичних нервів — переміщення в клітини калію відбувається шляхом стимуляції β2-адренергічних рецепторів [8]. У хворих на ЦД прийом неселективних β-блокаторів, підвищення адренергічної активності можуть посилити гіперкаліємію, оскільки, не зустрівши опору стимуляції α-рецепторів, сприяють виходу калію з клітини [12].
Кислотно-основні зміни. ДКА характеризується накопиченням ацетооцтової і β-оксимасляної кислот [14]. Кетоацидоз — це результат того, що швидкість, при якій утворюються печінкові кетокислоти, перевищує периферийну утилізацію та їх концентрація в крові збільшується, а накопичення протонів у позаклітинній рідині призводить до розкладання бікарбонату натрію на вуглекислий газ та воду, тоді як концентрація аніонів кетокислот зростає. Зниження концентрації бікарбонату в плазмі спочатку є пропорційним збільшенню аніонного розриву [27]. Хоча органічний ацидоз є домінантним порушенням при ДКА, мінеральний ацидоз також може відмічатися залежно від стадії захворювання [1, 11, 40] (рис. 2).
/16-26/19-1.jpg)
На ранній стадії ДКА, коли об’єм позаклітинної рідини (ОПР) близький до норми, то аніони кетокислот будуть швидко виводитися з організму нирками у вигляді солей натрію та калію. Діуретичні втрати кетонових солей призводять до зниження ОПР і сигналізують про ниркову затримку дієтичного (харчового) хлориду натрію. Протон кетокислот реагує з бікарбонатом, утворюючи воду та вуглекислий газ, що вивільняється через легені. Чистий ефект спостерігається при мінеральному ацидозі. Цей процес називається непрямою втратою бікарбонату натрію [27]. У міру того як кетогенез стає більш прискореним, а волемічне виснаження тяжчим, велика частина генерованих солей кетокислот зберігається в організмі, тим самим збільшуючи аніон-розрив. У цей момент швидкість клубочкової фільтрації (ШКФ), як правило, зменшується, і пацієнт потребує лікування та госпіталізації. Під час фази відновлення метаболічний ацидоз з аніон-розривом (органічний ацидоз) знову перетворюється в гіперхлоремічний ацидоз із нормальним аніон-розривом (мінеральний ацидоз). Лікування призводить до припинення продукції кетокислот. У результаті відновлення ОПР збільшується ниркова екскреція натрієвих солей з аніонів кетокислот. Непрямі втрати бікарбонату в поєднанні зі збереженням введеного натрію хлориду пояснюють перехід з органічного в мінеральний ацидоз. Крім того, калій і натрій входять до складу розчинів, що містять хлориди натрію і калію, які в клітинах обмінюються на іони водню. Відмічається чистий ефект вливання хлориду водню в позаклітинне середовище. Нормалізація кислотно-лужного балансу здійснюється протягом декількох днів, оскільки коригування дефіциту бікарбонату відбувається шляхом його регенерації нирками.
Ресусцитація збалансованими розчинами електролітів може зменшити мінеральний ацидоз під час фази відновлення [34]. Слід відмітити і той факт, що в пацієнтів із ДКА нирки первинно не залучаються (при їх нормальній функції). Нирки компенсують шляхом високих виділень амонію. Коли є втрата аніонів органічних кислот, то кількість амонію в сечі може бути оцінена шляхом вимірювання осмолярного діурезу, що визначається за рівнянням: сечова осмолярність = ([2 • Na+ сечі] + [2 • К+ сечі]) + + (азот сечовини в мл/дл / 2,8) + (глюкоза в сечі в мл/дл / 18).
За відсутності глюкозурії частина рівняння, що стосується глюкози, опускається. При збільшенні екскреції амонію в поєднанні з хлорид- або кето–аніонами спостерігається значне збільшення сечової осмолярної різниці — процес, типовий для ацидозу екстраренального характеру при збереженій функції нирок. Проте в деяких пацієнтів із ДКА, при інтактних нирках відповідь на кислотне навантаження нижча. Така відповідь відмічатиметься в пацієнтів із низькою ШКФ, коли знижується відфільтроване навантаження натрію. Рівноцінне зниження витрат аденозинтрифосфату, необхідне для транспорту калію та окислення кетоаніонів у клітинах проксимальних канальців, викликає зниження утилізації глутаміну і, отже, обмежує швидкість амоніогенезу [18, 21]. Вимірювання сечової осмолярної різниці є найбільш корисним при оцінці пацієнтів із мінеральним ацидозом (ця умова, як правило, ураховується під час фази відновлення ДКА після нормалізації аніонного розриву). Сечовий осмолярний розрив вимірюється для того, щоб вияснити, чи існує відповідне збільшення екскреції амонію, що призводить до розладу кислотно-основного стану. Низький сечовий осмолярний розрив у пацієнтів зі стійким гіперхлоремічним ацидозом передбачає канальцеву дисфункцію.
У минулому для визначення кетокислот використовувалися таблетки нітропрусиду або реагент-смужки. Незважаючи на недавні успіхи в дослідженні, що дозволили пряму оцінку рівня β-оксибутирату, визначення концентрації кетонових тіл часто не може повністю пояснити збільшення аніон-розривів, що, зі свого боку, у багатьох пацієнтів призводить до гіперлактатемії [10]. Рівень лактату може збільшуватись у відповідь на підвищення адренергічної активності, навіть за відсутності гіпоперфузії тканин [6].
Іншою причиною збільшення розриву є накопичення D-лактату, викликане збільшенням продукції метилгліоксалю в патологічному циклі гліоксалю. Незважаючи на такі фактори, як ацидоз, гіперосмолярність і церебральна гіпоперфузія, що виникають у пацієнтів із ДКА, є припущення, що D-лактат може також мати певний вплив [39]. Хронічна гіперглікемія призводить до розвитку карбонільного стресу та накопичення в організмі токсичних дикарбонільних сполук (метилгліоксалю, гліоксалю, 3-деоксиглюкозону та ін.) [25], наприклад метилгліоксалю, що збільшує утворення кінцевих продуктів глікозування білків, а NO гальмує їх накопичення. Дикарбонільні сполуки — це високореактивні продукти, що глікозують білки, ліпіди, нуклеїнові кислоти, що, зі свого боку, призводять до утворення кінцевих продуктів глікозування. Такий стан має назву «карбонільний стрес». Глікозовані білки –роблять внесок у розвиток атеросклеротичного пошкодження судин, виникнення мікроангіопатій, нефропатій та нейропатій. Вони стимулюють секрецію запальних цитокінів, агрегацію тромбоцитів, хемотаксис моноцитів, експресію ендотеліального фактора росту судин тощо [57].
Лікування ДКА включає в себе введення інсуліну та внутрішньовенне введення рідини для регідратації. Застосування олужнюючих розчинів зазвичай не потрібне, оскільки інсулін знижує швидкість утворення кетокислот, а окислення кетоаніонів веде до утворення бікарбонату. Але є певні ситуації, при яких застосовують лужні розчини, зокрема при високому ризику розвитку набряку головного мозку в дітей [9, 27].
Метморфін, що використовується при лікування ЦД 2-го типу, може в окремих випадках призвести до лактат-ацидозу, але ризик досить низький і насправді нічим не відрізняється від швидкості лактат-ацидозу в пацієнтів на фоні ЦД 2-го типу [26]. Ризик зростає, коли різко знижується функція нирок. Оскільки метморфін виводиться саме нирками, то він підвищується при порушенні їх функції, а саме при різкому зменшенні ШКФ, що може призвести до лактат-ацидозу [47]. Нестероїдні протизапальні препарати можуть збільшити ризик накопичення метформіну, оскільки звужують аферентні артеріоли, особливо в пацієнтів із зниженою нирковою перфузією, тим самим спричиняють різке та вагоме зменшення ШКФ. Метморфін легко видаляється за допомогою діалізної терапії (має низьку молекулярну масу та не зв’язує білки). Однак, щоб знизити рівень метморфіну, як правило, необхідна подовжена екстракорпоральна терапія, оскільки вона передбачає великий розподіл і двокамерну кінетичну елімінацію [31, 53].
Гіперкаліємічний ренальний тубулярний ацидоз. Гіперкаліємічний ацидоз ниркових канальців, або нирковий тубулярний ацидоз 4-го типу (НТА4т), є загальною ознакою серед пацієнтів із ЦД і вираженою нефропатією. Хвороба характеризується порушеннями функції нефронів, що призводить до порушення ниркової екскреції водню та калію з розвитком у подальшому гіперкаліємії та гіперхлоремічного (мінерального) ацидозу (рис. 3).
/16-26/21-1.jpg)
У нормальних умовах реабсорбція натрію в збірній трубці (канальці), по якій рухається альдостерон, генерує від’ємний потенціал у просвіті, що є рушійною силою для секреції калію в основних клітинах та іонів водню в α-інтеркальованих клітинах. Ослаблення реабсорбції натрію в основних клітинах призводить до зменшення електронегативності в просвіті збірної трубки. Це може відбуватися за рахунок зниження функції збірних канальців або спричинене гіпоренінемічним гіпоальдостеронізмом. Це веде до зменшення порушення секреції калію й іонів водню, сприяючи гіперкаліємії та метаболічному ацидозу. Гіперкаліємія також погіршує окислення за рахунок зменшення кількості амонію, здатного виступати як сечовий буфер. По-перше, гіперкаліємія знижує секрецію амонію в проксимальних канальцях. Точний механізм, за допомогою якого це відбувається, достеменно невідомий, але може включати в себе вхід калію до клітини в обмін на протони, що збільшують внутрішньоклітинний рН. По-друге, транспортування амонію по товстій висхідній гілці гальмується значним збільшенням концентрації калію в просвіті, що ефективно конкурує з амонієм для транспорту натрій-калій-хлорид котранспортера. Амоній зазвичай виходить із базолатеральної поверхні клітини через натрій-протонний обмінник 4. Рівень отриманих у результаті екскреції кислот зменшується через обмежену кількість буфера в поєднанні зі зниженням здатності секреція іонів водню. Діуретичний осмолярний розрив не збільшується, що вказує на те, що екскреція амонію в сечі є незначною чи відсутня. Пацієнти, у яких НТА4т викликаний дефектом мінералокортикоїдної активності, як правило, мають рН сечі менше 5,5, що відображає більш серйозний дефект за наявності амонію, аніж секреції іонів водню. рН сечі у хворих із структурними ушкодженнями більш лужний, ніж у пацієнтів із порушеною мінералокортикоїдною активністю, оскільки секреція іонів водню ослаблюється по всій збірній трубці (корковому та мозковому сегментах).
НТА4т може відмічатися в пацієнтів навіть із легким та середнім ступенем тяжкості діабетичної нефропатії, рівні гіперкаліємії та ацидозу можуть бути непропорційно значними щодо відстежених знижень ШКФ. Дефіцит альдостерону чи захворювання, що вражають збірні канальці, можуть призвести до НТФ4т, що, зі свого боку, веде до дефекту секреції іонів водню в дистальних канальцях [28]. Фактично в таких випадках множинні зміни в ренін-–ангіотензивній системі вагомо знижують циркуляцію чи активність мінералокортикостероїдів. Гіпоренінемічний гіпоальдостеронізм і волемічне зростання наявні в багатьох пацієнтів із ЦД. Вважають, що вони викликані зменшенням ШКФ через розвиток діабетичної нефропатії, та дослідження на тваринах вказують на те, що прямі ефекти інсуліну на рецептори в дистальному нефроні знижують активність лізиндефіцитної протеїнкінази-4, що призводить до патологічних змін, аналогічні спостерігаються при синдромі сімейної гіперкаліємічної артеріальної гіпертензії (псевдогіпоальдостеронізм 2-го типу) [23, 54].
Більшість пацієнтів не потребують лікування НТА4т при умові, що не мають одночасно захворювання, що посилює гіперкаліємію та ацидоз. Тому першочерговою метою терапії є корекція гіперкаліємії. У багатьох випадках зниження концентрації калію в плазмі буде нейтралізувати ацидоз. Терапією першої лінії є відмова від ліків, що втручаються в синтез чи активність альдостерону [44]. У пацієнтів із дефіцитом альдостерону, які не мають ані гіпертонії, ані перевантаження рідиною, введення синтетичних мінералокортикостероїдів (наприклад, флудрокортизону) є ефективним. У більшості пацієнтів з артеріальною гіпертензією чи в пацієнтів із передбачуваною ШКФ < 30 мл/хв у петлі Генле введення тіазидних сечогінних препаратів є альтернативним варіантом [44]. Введення від 30 до 40 ммоль бікарбонату на день, як правило, є достатнім, щоб підняти рівень бікарбонатів вище 20 ммоль/л у пацієнтів із персистуючим метаболічним ацидозом. Первинне ускладнення, пов’язане з такою терапією, — це перевантаження об’єму, хоча утримання натрію за допомогою бікарбонату натрію є нижчим, ніж хлоридом натрію в пацієнтів із хронічним захворюванням нирок [24]. При лікуванні НТА4т у хворих на ЦД виникає терапевтична дилема щодо вибору препаратів, що блокують ренін-ангіотензивну систему. Незважаючи на те, що такі препарати, як інгібітори ангіотензинперетворюючого ферменту чи блокатори рецепторів ангіотензину є корисними для сповільнення прогресування захворювання нирок та лікування серцевої недостатності, вони можуть також збільшити ризик розвитку небезпечної для життя гіперкаліємії.
Двовалентні катіони та фосфор. Зміни в гомеостазі двовалентних катіонів і фосфору пов’язані з гіперглікемією, тому часто спостерігаються в пацієнтів із ЦД. Епідеміологічні дослідження показують, що низький рівень споживання магнію пов’язаний із підвищеним ризиком розвитку діабету, тоді як більш високе споживання магнію — із низьким ризиком розвитку діабету [55]. Крім того, гіпомагніємія може призвести до порушення утилізації глюкози та сприяти серцево-судинним захворюванням, ретинопатії та нефропатії [4]. Випадки гіпомагніємії у хворих з 2-м типом ЦД становлять 13,5–47,7 % [49]. Причинами цього є недостатній пероральний прийом та хронічна діарея, пов’язана з вегетативною нейропатією. Інгібітори протонного насоса знижують всмоктування магнію з шлунково-кишкового тракту. Цей ефект може бути результатом медикаментозного зниження рН в просвіті кишки, що призводить до зміни спорідненості перехідного потенціалу рецепторів каналів меластатин-6 та меластатин-7 на апікальній поверхні ентероцитів до магнію [48].
У пацієнтів із ДКА осмотичний діурез через поганий глікемічний контроль призводить до ниркових втрат магнію. Проте сироватковий рівень магнію може бути дещо підвищеним унаслідок дефіциту інсуліну та метаболічного ацидозу, незважаючи на загальне виснаження організму. Зміни магнію в клітинах при введенні інсуліну та корекції ацидозу призводять до викриття дефіциту в організмі. Крім того, збільшення активності адренорецепторів можуть сприяти внутрішньоклітинним змінам магнію [50].
Гіпокальціємія є потенційним ускладненням діабетичної нефропатії у хворих із нефротичним синдромом, оскільки нефротичний стан призводить до втрати сечового 25-гідроксивітаміну D3 та його протеїнового носія [17].
Зміни показника паратгормона у хворих на ЦД такі, як при гіпопаратиреозі, і мають тенденцію до розвитку гіпокальціємії [35, 52]. Гіпомагнезіємія може бути причиною гіпокальціємії, оскільки дефіцит магнію призводить до зниження вивільнення паратиреоїдного гормона. Первинний гіперпаратиреоз слід розглядати в пацієнтів із ЦД, які мають гіперкальціємію, оскільки первинний гіперпаратиреоз розвивається в них значно швидше в кілька разів [56]. Гіперкальціємія розвивається також у пацієнтів із дегідратацією, що призводить до підвищеної ниркової реабсорбції кальцію [45].
Розлад фосфатного метаболізму спостерігається в пацієнтів із ДКА. Гіперфосфатемія часто наявна при рівні вище від 17 мг/дл через дефіцит інсуліну і метаболічний ацидоз [15]. Лікування інсуліном та корекція ацидозу викликають різке зниження концентрації фосфору в плазмі за рахунок зрушення в клітинах, дефіцит становить 1 ммоль/кг маси тіла [30, 45]. Зниження концентрації фосфору в плазмі є також результатом сечових втрат, викликаних осмотичним діурезом. За відсутності ДКА гіперфосфатемія наявна при гострому пошкодженні чи переході в хронічне захворювання нирок (що також спостерігається в пацієнтів без діабету).
Рандомізовані дослідження фосфатної терапії у хворих із ДКА не встановили, що вона забезпечує клінічний ефект. Таким чином, рутинне введення фосфату не рекомендоване [13]. Ризик розвитку потенційного ускладнення гіпофосфатемії відмічається в пацієнтів із серцевою слабкістю чи слабкістю скелетних м’язів, рабдоміолізом, гемолітичною анемією, тому калій фосфат може бути доданий до інфузійних розчинів. Звичайно, гіпокальціємія та гіпомагніємія є потенційними ускладненнями введення фосфату (табл. 1).
Таким чином, порушення регуляції гомеостазу глюкози призводить до великої кількості прямих і непрямих впливів на водно-електролітний і кислотно-лужний баланс, оскільки велика поширеність діабету гарантує, що лікарі практично кожної спеціальності мають справу з пацієнтами, хворими на цукровий діабет, а знання, пов’язані зі змінами електролітів, є необхідними.
Список литературы
1. Adrogué H.J., Eknoyan G., Suki W.K. Diabetic ketoacidosis: role of the kidney in the acid-base homeostasis re-evaluated // Kidney Int. — 1984. — 25. — 591-8.
2. Alterations in nonenzymatic biochemistry inuremia: origin and significance of «carbonyl stress» in long-term uremic complications / [T. Miyata, C. van Ypersele de Strihou, K. Kurokawa et al.] // Kidney Int. — 1999. — № 2. — P. 389-399.
3. Aronson P.S., Giebisch G. Effects of pH on potassium: new explanations for old observations // J. Am. Soc. Nephrol. — 2011. — 22. — 1981-9.
4. Barbagallo M., Dominguez L.J. Magnesium metabolism in type 2 diabetes mellitus, metabolic syndrome and insulin resistance // Arch. Biochem. Biophys. — 2007. — 458. — 40-7.
5. Beukhof C.M., Hoorn E.J., Lindemans J., Zietse R. Novel risk factors for hospital ac-quired hyponatraemia: a matched casecontrol study // Clin. Endocrinol. (Oxf.). — 2007. — 66. — 367-72.
6. Bolli G., Cartechini M.G., Compagnucci P. et al. Adrenergic activity and glycometa-bolic compensation in patients with diabetes mellitus // Minerva Med. — 1979. — 70. — 3783-95.
7. Bustamante M., Hasler U., Kotova O. et al. Insulin potentiates AVP-induced AQP2 expression in cultured renal collecting duct principal cells // Am. J. Physiol. Renal. Physiol. — 2005. — 288. — 334-344.
8. Christensen N.J. Plasma norepinephrine and epinephrine in untreated diabetics, during fasting and after insulin administration // Diabetes. — 1974. — 23. — 1-8.
9. Chua H.R., Schneider A., Bellomo R. Bicarbonate in diabetic ketoacidosis — a systematic review // Ann. Intensive Care. — 2011. — 1. — 23.
10. Cox K., Cocchi M.N., Salciccioli J.D., Carney E., Ho–well M., Donnino M.W. Prevalence and significance of lactic acidosis in diabetic ketoacidosis // J. Crit. Care. — 2012. — 27. — 132-7.
11. Elisaf M.S., Tsatsoulis A.A., Katopodis K.P., Siamopoulos K.C. Acid-base and electrolyte disturbances in patients with diabetic ketoacidosis // Diabetes Res. Clin. Pract. — 1996. — 34. — 23-7.
12. Epstein F.H., Rosa R.M. Adrenergic control of serum potassium // N. Engl. J. Med. — 1983. — 309. — 1450.
13. Fisher J.N., Kitabchi A.E. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis // J. Clin. Endocrinol. Metab. — 1983. — 57. — 177-80.
14. Foster D.W., McGarry J.D. The metabolic derangements and treatment of diabetic ketoacidosis // N. Engl. J. Med. — 1983. — 309. — 159-69.
15. Franks M., Berris R.F. et al. Metabolicstudies in diabetic acidosis; the effect of the administration of sodium phosphate // Arch. Intern. Med. (Chic.). — 1948. — 81. — 42-55.
16. Goguen J., Gilbert J. Hyperglycemic emergencies in adults // Can. J. Diabetes. — 2013. — 37. — 72-76.
17. Goldstein D.A., Haldimann B., Sherman D., Norman A.W., Massry S.G. Vitamin D metabolites and calcium metabolism in patients with nephrotic syndrome and normal renal function // J. Clin. Endocrinol. Metab. — 1981. — 52. — 116-21.
18. Halperin M.L., Cheema-Dhadli S. Renal and hepatic aspects of ketoacidosis: a quantitative analysis based on energy turno–ver // Diabetes Metab. Rev. — 1989. — 5. — 321-36.
19. Halperin M.L., Kamel K.S. Some observations on the clinical approach to metabolic acidosis // J. Am. Soc. Nephrol. — 2010. — 21. — 894-7.
20. Halperin M.L., Margolis B.L., Robinson L.A., Halperin R.M, West M.L., Bear R.A. The urine osmolal gap: a clue to estimateurine ammonium in «hybrid» types of metabolic acidosis // Clin. Invest. Med. — 1988. —11. — 198-202.
21. Halperin M.L., Vinay P., Gougoux A., Pichette C., Jungas R.L. Regulation of the maxi-mum rate of renal ammoniagenesis in the acidotic dog // Am. J. Physiol. — 1985. — 248. — 607-615.
22. Hillier T.A., Abbott R.D., Barrett E.J. Hyponatremia: evaluating the correction factor for hyperglycemia // Am. J. Med. — 1999. — 106. — 399-403.
23. Hoorn E.J., Nelson J.H., McCormick J.A., Ellison D.H. The WNK kinase network regulating sodium, potassium, and blood pressure // J. Am. Soc. Nephrol. — 2011. — 22. — 605-14.
24. Husted F.C., Nolph K.D., Maher J.F. NaHCO3 and NaC1 tolerance in chronic renal failure // J. Clin. Invest. — 1975. — 56. — 414-9.
25. Hyperglycemia and glycation in diabetic complications / [A. Fegre-Salvayre, R. Salvayre, N. Augé et al.] // Antioxid. Redox Signal. —2009. — № 11. — P. 3071-3109.
26. Inzucchi S.E., Lipska K.J., Mayo H., Bailey C.J., –McGuire D.K. Metformin in patients with type 2 diabetes and kidney disease: a systematic review // JAMA. — 2014. — 312. — 2668-75.
27. Kamel K.S., Halperin M.L. Acid-base problems in diabetic ketoacidosis // N. Engl. J. Med. — 2015. — 372. — 546-54.
28. Karet F.E. Mechanisms in hyperkalemic renal tubular acidosis // J. Am. Soc. Nephrol. — 2009. — 20. — 251-4.
29. Kebler R., McDonald F.D., Cadnapaphornchai P. Dynamic changes in serum phosphorus levels in diabetic ketoacidosis // Am. J. Med. — 1985. — 79. — 571-6.
30. Kitabchi A.E., Umpierrez G.E., Miles J.M., Fisher J.N. Hyperglycemic crises in adult patients with diabetes // Diabetes Care. — 2009. — 32. — 1335-43.
31. Lalau J.D., Andrejak M., Morinière P. et al. Hemodialysis in the treatment of lactic acidosis in diabetics treated by metformin: a study of metformin elimination // Int. J. Clin. Pharmacol. Ther. Toxicol. — 1989. — 27. — 285-8.
32. Liamis G., Liberopoulos E., Barkas F., Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians // Am. J. Nephrol. — 2013. — 38. — 50-7.
33. Lu J., Zello G.A., Randell E., Adeli K., Krahn J., Meng Q.H. Closing the anion gap: contribution of D-lactate to diabetic ketoacidosis // Clin. Chim. Acta. — 2011. — 412. — 286-91.
34. Mahler S.A., Conrad S.A., Wang H., Arnold T.C. Resuscitation with balanced electrolyte solution prevents hyperchloremic metabolic acidosis in patients with diabetic ketoacidosis // Am. J. Emerg. Med. — 2011. — 29. — 670-4.
35. McNair P., Christensen M.S., Madsbad S., Christiansen C., Transbøl I. Hypoparathyroidism in diabetes mellitus // Acta Endocrinol. (Copenh.) — 1981. — 96. — 81-6.
36. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. — Atlanta: Centers for Disease Control and Prevention, 2014.
37. Nguyen T.Q., Maalouf N.M., Sakhaee K., Moe O.W. Comparison of insulin action on glucose versus potassium uptake in humans // Clin. J. Am. Soc. Nephrol. — 2011. — 6. — 1533-9.
38. Nyenwe E.A., Kitabchi A.E. Evidencebased management of hyperglycemic emergencies in diabetes mellitus // Diabetes Res. Clin. Pract. — 2011. — 94. — 340-51.
39. Nyenwe E.A., Razavi L.N., Kitabchi A.E., Khan A.N., Wan J.Y. Acidosis: the prime determinant of depressed sensorium in diabetic ketoacidosis // Diabetes Care. — 2010. — 33. — 1837-9.
40. Oh M.S., Carroll H.J., Uribarri J. Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis // Nephron. — 1990. — 54. — 1-6.
41. Palmer B.F., Gates J.R., Lader M. Causes and management of hyponatremia // Ann. Pharmacother. — 2003. — 37. — 1694-702.
42. Palmer B.F. A physiologic-based approach to the evaluation of a patient with hyperkalemia // Am. J. Kidney Dis. — 2010. — 56. — 387-93.
43. Palmer B.F. A physiologic-based approach to the evaluation of a patient with hypokalemia // Am. J. Kidney Dis. — 2010. — 56. — 1184-90.
44. Palmer B.F. Managing hyperkalemia caused by inhibitors of the rennin-angiotensin-aldosterone system // N. Engl. J. Med. — 2004. — 351. — 585-92.
45. Palmer B.F. Metabolic complications associated with use of diuretics // Semin. Nephrol. — 11. — 31. — 542-52.
46. Palmer B.F. Regulation of potassium homeostasis // Clin. J. Am. Soc. Nephrol. — 2015. — 10. — 1050-60.
47. Palmer B.F. Renal dysfunction complicating the treatment of hypertension // N. Engl. J. Med. — 2002. — 347. — 1256-61.
48. Perazella M.A. Proton pump inhibitors and hypomagnesemia: a rare but serious complication // Kidney Int. — 2013. — 83. — 553-6.
49. Pham P.C., Pham P.M., Pham S.V., Miller J.M., Pham P.T. Hypomagnesemia in patients with type 2 diabetes // Clin. J. Am. Soc. Nephrol. — 2007. — 2. — 366-73.
50. Romani A.M. Cellular magnesium homeostasis // Arch. Biochem. Biophys. — 2011. — 512. — 1-23.
51. Roscoe J.M., Halperin M.L., Rolleston F.S., Goldstein M.B. Hyperglycemia-inducedhyponatremia: metabolic considerations in calculation of serum sodium depression // Can. Med. Assoc. J. — 1975. — 112. — 452-3.
52. Schwarz P., Sørensen H.A., Momsen G., Friis T., Transbøl I., McNair P. Hypocalcemia and parathyroid hormone responsiveness in diabetes mellitus: a tri-sodiumcitrate clamp study // Acta Endocrinol. (Copenh.). — 1992. — 126. — 260-3.
53. Seidowsky A., Nseir S., Houdret N., Fourrier F. Metformin-associated lactic acidosis: a prognostic and therapeutic study // Crit. Care Med. — 2009. — 37. — 2191-6.
54. Song J., Hu X., Riazi S., Tiwari S., Wade J.B., Ecelbar–ger C.A. Regulation of blood pressure, the epithelial sodium channel (ENaC), and other key renal sodium transporters by chronic insulin infusion in rats // Am. J. Physiol. Renal. Phy–siol. — 2006. — 290. — 1055-1064.
55. Song Y., Manson J.E., Buring J.E., Liu S. Dietary magnesium intake in relation to plasma insulin levels and risk of type 2 diabetes in women // Diabetes Care. — 2004. — 27. — 59-65.
56. Taylor W.H., Khaleeli A.A. Coincident diabetes mellitus and primary hyperparathyroidism // Diabetes Metab. Res. Rev. — 2001. — 17. — 175-80.
57. Turk Z. Glycotoxines, carbonyl stress and relevance to –diabetes and its complications / Z. Turk // Physiol. Res. — 2010. — № 2. — P. 147-156.
58. Tzamaloukas A.H., Ing T.S., Elisaf M.S. et al. Abnormalities of serum potassium concen-tration in dialysis-associated hyperglycemia and their correction with insulin: a unique cli-nical/physiologic exercise in internal potassium balance // Int. Urol. Nephrol. — 2010. — 42. — 1015-22.

/16-26/18-1.jpg)
/16-26/19-1.jpg)
/16-26/21-1.jpg)
/16-26/22-1.jpg)
/16-26/23-1.jpg)