Ізольований (селективний) дефіцит IgE — одна з найпоширеніших у людській популяції первинних імунодефіцитних хвороб, що характеризується кількісним і/або якісним дефектом молекули імуноглобуліну класу Е за нормальних інших відомих компонентів імунної системи [2]. Відноситься до мінорних імунодефіцитів, або малих імунних дисфункцій. Може бути тотальним і парціальним. Має варіабельний перебіг і гетерогенну клінічну картину, що включає інфекційні, алергічні, псевдоалергічні, автоімунні, імунозапальні, онкологічні та деякі додаткові прояви, у тому числі персистуючу фіброміалгію і синдром хронічної втоми [12, 13, 24]. Механізми розвитку клінічних проявів цього імунодефіциту добре систематизовані M.B. Pate зі співавт. (рис. 1). У даний час встановлено, що частота цієї імунодефіцитної хвороби в загальній популяції становить один випадок на 30 осіб, однак може бути вищою серед пацієнтів з імунозалежною патологією, що звертаються по медичну допомогу до лікувально-профілактичних закладів [29]. Хоча ця імунна дисфункція була описана вперше ще в 1969 році V.A. Cain зі співавт. [10] та A.J. Ammann зі співавт. [7] незалежно одні від одних, лікарі країн Східної Європи досі дуже погано поінформовані з цієї проблеми. Тому раніше ми опублікували всебічний огляд наукових статей з проблеми клініки, діагностики й лікування ізольованого дефіциту IgE у людей [2]. Відповідна доповідь була викладена на VI Світовому конгресі з астми і хронічної обструктивної хвороби легень (Лондон, 2013) [15]. Ці кроки мали на меті звернути увагу медичної спільноти на необхідність раціональної діагностики й лікування цієї поширеної імунодефіцитної хвороби. Однак протягом останніх років були накопичені нові дані, які розширюють сучасні уявлення щодо природи вибіркового дефіциту IgE у людей, що спонукало нас до підготовки додаткової публікації з цієї проблеми.
/9-1.gif)
По-перше, зазнали уточнення фундаментальні погляди на біологічне призначення молекул IgE в організмі людини. Згідно із загальновизнаною концепцією Джексона Коллінза, що описує й пояснює послідовність синтезу імуноглобулінів різних класів під час гострої імунної відповіді, IgE є важливою складовою ранньої, негайної фази імунного запалення, під час якої функціонує в тісному взаємозв’язку з молекулами IgМ. При цьому низькоафінні тканинні IgE завдяки індукції активації й дегрануляції тучних клітин слизових оболонок вхідних воріт інфекції й розвитку пов’язаної з цим локальної захисної ексудативної реакції критично зменшують абсорбцію антигенів із вогнища первинного афекту до кровотоку, а циркулюючі молекули IgE видаляють решти антигену, що проникли до мікроциркуляторного русла, до периферичних тканин, шляхом активації базофілів крові. Такий механізм обмежує вогнище запалення і запобігає генералізації інфекції [3].
Справді, сьогодні все більше публікацій присвячено вивченню біологічних функцій молекул IgE в організмі людини. Без сумніву, основним призначенням цього класу антитіл є індукція ексудативної запальної реакції, яка важлива в захисті від багатьох інфекцій та інвазій, однак незамінна — при кишкових нематодозах. Іншими антигензалежними функціями IgE є нейтралізація антигенів і токсинів, опсонізація й участь у реакціях антитілозалежної клітинно-опосередкованої цитотоксичності, здебільшого — при тканинних формах гельмінтів, наприклад трихінельозі. До альтернативних, або антигеннезалежних, функцій IgE відноситься регуляція секреторної активності й витривалості тучних клітин [14], експресії молекул на їх поверхні та взаємодії з фібронектином міжклітинного матриксу [17].
При осмисленні критеріїв діагностики дефіциту IgE слід виходити з розуміння факту, що застосовувані нині методики імуноферментного аналізу для верифікації діагнозу гуморальної імунної дисфункції дають передусім відносні, а не абсолютні результати [1]. Класичні критерії діагностики ізольованого дефіциту IgE у людей запропоновані J.J. Schoettler зі співавт. у 1989 році. Згідно з цими критеріями тотальний дефіцит IgE діагностують при зниженні сироваткової концентрації IgE до рівня 10 МО/мл, а тотальний дефіцит — до 5 МО/мл і нижче [28]. Зазначені критерії ґрунтуються на діагностичних можливостях традиційних методик імуноферментного аналізу, що застосовуються в даний час в Україні найчастіше. Крім того, у деяких діагностичних центрах доступні методики так званого високоточного імуноферментного аналізу, згідно з результатами якого діагноз дефіциту IgE констатують при зниженні сироваткової концентрації імуноглобуліну цього класу менше від 2 МО/мл. Нещодавно F. Famuyiwa, I. Rubinstein розробили спеціальну методику ультраcенситивного високоточного імуноферментного аналізу, при застосуванні якої дефіцит IgE можна діагностувати при зниженні сироваткової концентрації до 0,2 МО/мл, однак такі реактиви у даний час недоступні для широкої клінічної практики і застосовуються здебільшого з науковою метою [11].
Завдяки генетичним дослідженням S. Roa зі спів–авт. стала відомою основна, хоча і не єдина причина первинного ізольованого дефіциту IgE у людей [27]. Як відомо, мутації в гені активаційно індукованої цитидиндезамінази (activation-induced cytidine deaminase, AICDA) В-лімфоцитів зумовлюють розвиток так званого гіпер-IgM-синдрому 2-го типу, при якому порушений процес переключення ізотипів антитіл під час імунної відповіді, який отримав назву рекомбінації класового переключення (class switch recombination, CSR). Відомі мутації AICDA, що лежать в основі гіпер-IgM-синдрому, призводять до проксимального порушення рекомбінативних процесів, що зумовлює надмірну продукцію ініціального класу IgM і виражений дефіцит дистальних ізотипів IgD, IgG, IgA та IgE (рис. 2).
/10-1.gif)
Завдяки методам конформаційного сенситивного електрофорезу в гелі та ДНК-секвенуванню S. Roa зі співавт. встановили, що заміни нуклеотидів 5923A/G і 7888C/T в гені AICDA В-клітин зумовлюють подібне, однак дистальне порушення механізму переключення ізотипів антитіл, при якому уражена тільки продукція завершального ізотипу IgE за нормального вироблення імуноглобулінів інших класів, що розташовуються проксимальніше в ланцюгу послідовності синтезу імуноглобулінів різних класів під час імунної відповіді [27]. Це є основою генетичної природи первинного ізольованого дефіциту IgE у людей, і в даний час визначення обох причинних поліморфізмів гену AICDA доступне в Україні, що полегшує діагностику імунодефіциту в клінічній практиці.
Останніми роками здійснено кілька нових епідеміо–логічних досліджень з вивчення частоти ізольованого дефіциту IgE у людей. Так, S. McVicker, M.Y. Karim виявили тотальний ізольований дефіцит IgE в 1,84 % випадків серед 4013 пацієнтів з алергологічною клінікою, що проходили процедуру вимірювання сироваткової концентрації IgE для підтвердження атопічної природи алергічного ураження. Крім того, автори показали, що в деяких випадках аномально низька концентрація IgE в алергологічній практиці може вказувати на ширшу імунодефіцитну хворобу — загальний варіабельний імунодефіцит, що потребує подальшого проведення розгорнутих імунологічних досліджень [23]. Нарешті проведено національне епідеміологічне дослідження, присвячене вивченню частоти ізольованого дефіциту IgE серед пацієнтів з імунозалежною патологією, що звертаються по медичну допомогу до клінічного імунолога і/або алерголога (n = 4476). У цілому ізольований дефіцит IgE в окресленій когорті пацієнтів зустрічався в 7 % випадків, причому тотальна форма хвороби (< 5 МО/мл) мала місце щонайменше в 3 %, а парціальна (< 10 МО/мл) — у 4 % випадків. Це були особи з рецидивними інфекційними синусо-пульмональними ураженнями (63 %), гастроінтестинальним синдромом (13 %), автоімунними проявами (10 %), алергічними реакціями (7 %) та синдромом хронічної втоми (7 %) [16]. Доповідь вітчизняних учених щодо результатів цієї роботи на VIII Світовому конгресі з імунопатології, респіраторної алергії та астми (Дубай, ОАЕ, 2013) відзначена спеціальною нагородою журі, що вказує на високу зацікавленість наукових кіл проблемою ізольованого дефіциту IgE у людей [22].
Відкритим донедавна залишалося питання щодо зв’язку ізольованого дефіциту IgE з клінічним фенотипом. У зв’язку з цим E. Magen зі співавт. провели велике популяційне дослідження за участю 18 487 осіб загальної популяції. За результатами цього дослідження, тотальний ізольований дефіцит IgE (< 2 kIU/L) зустрічався серед 318 учасників, що становило 1,7 % випадків. Лише третину пацієнтів становили діти віком від 4 до 12 років (30,9 %), а інші 250 осіб (69,1 %) перебували в дорослій віковій категорії. Продемонстровано тісний зв’язок фенотипу тотального ізольованого дефіциту IgE з бронхіальною астмою й гіперреактивністю бронхіального дерева в дітей, а також хронічним синуситом, середнім отитом, різноманітними автоімунними й онкологічними синдромами в обох вікових групах [20]. Результати цього великого добре спланованого епідеміологічного дослідження вказують на велику питому вагу цього імунодефіциту серед дорослих, що не відповідає класичним уявленням про переважання первинних імунодефіцитів серед дітей, а також уточнює частоту тотального варіанта хвороби серед осіб загальної популяції та вказує на зв’язок із гетерогенною клінічною симптоматикою, яка може погіршити якість життя або навіть стати причиною смерті пацієнтів. Ці дані змушують відповідальніше ставитися до проблеми своєчасної діагностики й лікування ізольованого дефіциту IgE у людей.
/10-2.gif)
Розширилися уявлення щодо механізмів розвитку мікробіндукованих ускладнень у пацієнтів із дефіцитом IgE. Нещодавно в контрольованому рандомізованому клінічному дослідженні за типом «випадок — конт–роль» продемонстровано зв’язок ізольованого дефіциту IgE зі шлунковою диспепсією, гастритом і пептичною виразкою, асоційованими з інфекцією, викликаною мікроорганізмом H. рylori. Досліджувану групу становили 156 осіб з ізольованим дефіцитом IgE віком понад 12 років, що були відібрані рандомізованим чином із великої когорти імуноскомпрометованих пацієнтів, які перебували під диспансерним спостереженням. Контрольна група була представлена 3160 особами загальної популяції. Інфекцію, викликану H. рylori, діагностували на підставі результатів дихального тесту. Статистичний аналіз отриманої інформації для виявлення зв’язків здійснювали за допомогою розрахунку критерію χ2 Пірсона й точного тесту Фішера. Хоча частота випадків шлункової диспепсії була однаковою в обох групах спостереження, позитивні результати тесту на H. рylori частіше зустрічалися саме серед пацієнтів з ізольованим дефіцитом IgE (74,4 проти 31,1 %; р < 0,01). Потреба в проведенні діагностичної фібро–езофагогастродуоденоскопії виникала вірогідно частіше серед імуноскомпрометованих пацієнтів (57,9 проти 29,8 %; р < 0,05). У досліджуваній групі частіше відзначалися випадки пептичної виразки шлунка порівняно з імунокомпетентними особами (63,2 проти 15,9 %; р < 0,01). Гістопатологічні дослідження демонстрували виражене лімфоцитарне запалення слизової оболонки, формування додаткових лімфоїдних фолікулів із вираженими гермінативними центрами, інфільтрацію шлункових залоз поліморфноядерними лейкоцитами. Крім того, серед пацієнтів із дефіцитом IgE частіше зустрічалися резистентні штами H. рylori, що вимагали проведення потрійної антибактеріальної терапії другого ряду (47,4 проти 11,7 %; р < 0,01). Автори дійшли висновку, що ізольований дефіцит IgE асоційований з вищою частотою H. pylori-індукованих гастриту й пептичної виразки шлунка та резистентністю до рекомендованих антибактеріальних ліків. Припускають, що в пацієнтів із дефіцитом IgE порушена захисна ексудативна реакція в слизовій оболонці шлунка у відповідь на H. рylori, яка, хоча і зумовлює картину транзиторної шлункової диспепсії при інфікуванні, однак не допускає проникнення мікроба в глиб слизової оболонки. У разі дефіциту IgE H. рylori легше здійснює інвазію в середину слизової оболонки, призводячи до активації клітинної імунної відповіді in situ, а значить, лімфоцитарної та нейтрофільної інфільтрації та деструкції шлункової слизової оболонки з формуванням картини гастриту й пептичної виразки [19]. Раніше повідомляли про зв’язок ізольованого дефіциту IgE з гастроезофагеальною рефлюксною хворобою [13].
Розширилися також уявлення щодо додаткових, неінфекційних проявів цього імунодефіциту. У рандомізованому контрольованому клінічному дослідженні за участю 103 пацієнтів досліджуваної і 1030 осіб контрольної групи вивчали зв’язок ізольованого дефіциту IgE з атеросклерозом і пов’язаними з ним ускладненнями. Показано, що в пацієнтів з ізольованим дефіцитом IgE частіше розвиваються артеріальна гіпертензія (34 (37,7 %) проти 187 (18,2 %); p < 0,001), ішемічна хвороба серця (26 (25,2 %) проти 87 (8,4 %); p < 0,001), каротидний стеноз (5 (4,9 %) проти 7 (0,7 %); p = 0,003), цереброваскулярна хвороба (3 (2,9 %) проти 5 (0,5 %); p = 0,029) і периферична васкулярна хвороба (4 (3,9 %) проти 9 (0,9 %); p = 0,024). Результати цього клінічного дослідження вказують на те, що дефіцит IgE, найбільш ймовірно, є незалежним чинником ризику розвитку атеросклерозу та пов’язаних із ним кардіо- і цереброваскулярних ускладнень, впливаючи на захворюваність і смертність пацієнтів третьої вікової групи. Автори припускають, що молекули IgE завдяки індукції захисної ексудативної реакції сприяють своєчасному видаленню антигенів зі стінки судин, запобігаючи пришвидшеному росту атеросклеротичних бляшок. Навпаки, дефіцит IgE спричиняє перевантаження бляшок антигенами, які циркулюють у крові, що зумовлює швидкий ріст атеросклеротичних уражень з індукцією їх нестабільності у зв’язку з розвитком місцевої клітинної запальної реакції в середині бляшки [18]. Крім того, результати нещодавніх епідеміологічних досліджень також указують на асоціацію ускладненого атеросклерозу артерій з іншою мінорною імунодефіцитною хворобою — первинним дефіцитом манозозв’язуючого білка, що уразив нині 5–10 % людської популяції [32].
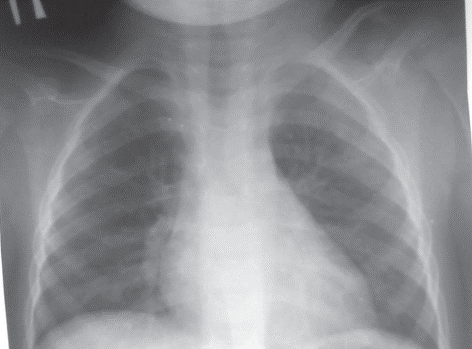
Завдяки повідомленням про клінічні випадки поповнилися знання щодо гетерогенності клінічної картини ізольованого дефіциту IgE у людей. Так, N. Riyaz зі співавт. описали розвиток у 27-річного пацієнта з ізольованим дефіцитом IgE автоімунних синдромів цукрового діабету 1-го типу та мікроспоричного коліту, імунозапального ураження шкіри pyoderma gangrenosum та ідіопатичного гіпереозинофільного синдрому без суттєвих проявів інфекційних уражень як на момент надходження до стаціонару, так і протягом анамнезу життя [26]. Також повідомляли про білатеральний інтерстиційний пневмоніт EBV-етіології в немовляти з миттєвою відповіддю на в/в імуноглобулін (рис. 4) та розсіяний склероз і лейоміому матки як ускладнення хронічної активної EBV-інфекції в дорослих з ізольованим дефіцитом IgE [2]. Крім того, повідомляли про кілька випадків тяжких герпесвірусних нейроінфекцій у пацієнтів із вибірковим дефіцитом IgE [21].
/12-1.gif)
Проведено ряд невеликих контрольованих клінічних досліджень, присвячених вивченню клінічного фенотипу імунодефіциту. Так, в одному контрольованому клінічному випробуванні продемонстровано зв’язок між парціальним і тотальним дефіцитом IgE і хронічним рефрактерним риносинуситом у людей [6]. В іншому контрольованому клінічному дослідженні показано часту асоціацію ізольованого дефіциту IgE з автоімунними демієлінізуючими хворобами периферичної нервової системи, включаючи синдром Гійєна — Барре й хронічну запальну демієлінізуючу полінейропатію, що дає додаткове пояснення високої ефективності імуноглобулінотерапії в таких випадках [4]. Раніше про подібну асоціацію повідомляли при іншому мінорному гуморальному імунодефіциті — ізольованому дефіциті субкласів IgG [9]. В іншому контрольованому клінічному дослідженні виявлено асоціацію ізольованого дефіциту IgE з рефрактерною тригемінальною невралгією, пов’язаною з реактивованими герпесвірусними інфекціями, включаючи викликані VZV, EBV і HHV-7 [5]. Ці дані дозволяють подивитися під іншим кутом на результати нещодавніх досліджень, які визначали клінічну ефективність в/в імуноглобулінотерапії при рефрактерних больових синдромах у людей [30]. Крім того, обговорено причинну роль дефіциту IgE у формуванні так званого холодного запалення в додаткових пазухах носа — феномену, який часто реєструється при проведенні магнітно-резонансного дослідження голови й нерідко викликає великі діагностичні труднощі в клініцистів [5]. Уперше про схильність до формування холодного запалення при ізольованому дефіциті IgE через брак ексудативної реакції повідомили S.H. Polmar зі співавт. у 1972 році [25].
Як відомо, для лікування ізольованого дефіциту IgE запропоновано препарати нормального імуноглобуліну людини для внутрішньом’язового [12] і внутрішньовенного введення [13]. У разі переважання бактеріальних інфекцій у клінічній картині імунодефіциту апробовано профілактичну антибіотикотерапію. Нещодавно опубліковані результати порівняльного контрольованого клінічного дослідження з вивчення ефективності двох доступних стратегій базисної імунотерапії клінічно маніфестного ізольованого дефіциту IgE. У цьому дослідженні брали участь 82 імуноскомпрометовані дорослі пацієнти віком від 16 до 61 року, причому 33 із них — тотальний дефіцит IgE (< 5 МО/мл), а в 49 — парціальна форма хвороби (< 10 МО/мл). Спочатку як базисну імунотерапію призначали препарат 10% нормального імуноглобуліну людини в дозі 0,3–0,4 мл/кг маси тіла болюсно кожні 3–4 тижні протягом 2–3 місяців поспіль, а в подальшому у пацієнтів, які не відповіли належним чином на стартову внутрішньом’язову імунотерапію, застосовували препарат 5% нормального імуноглобуліну людини для внутрішньовенного введення в дозі 200–400 мг/кг маси тіла 1 раз на 3–4 тижні ще протягом 1–3 місяців як другий ряд терапії. При цьому досягти усунення клінічних проявів імунодефіциту вдалося у 87 % випадків. Крім того, імунотерапія спричинила посилення ендогенної продукції молекул IgE — від 3,05 ± 1,21 МО/мл до 12,5 ± 1,86 МО/мл при тотальному дефіциті (р < 0,01) та від 7,23 ± 1,37 МО/мл до 15,66 ± 1,66 МО/мл при парціальній формі хвороби [31]. Таким чином, досягнуто не лише сурогатне заміщення, але й реалізовано імуномодулюючий ефект, що забезпечив потенціювання порушеної ланки імунітету. Раніше імуномодулюючий ефект препаратів імуноглобуліну при іншій імунодефіцитній хворобі — загальному варіабельному імунодефіциті — продемонстрували J. Bayry зі співавт., пояснивши отримані збільшення розмірів гермінативних центрів лімфатичних вузлів і посилення ендогенного антитілогенезу під впливом імунотерапії феноменом модуляції В-лімфоцитів через їх Fc-рецептори константними ділянками екзогенних молекул IgG препарату [8]. Подібно до цього автори даного порівняльного дослідження пояснили досягнутий імуномодулюючий ефект застосовуваної ступінчастої імунотерапії стимулюючим впливом введених молекул IgG препаратів на Fc-рецептори В-клітин, комітованих до продукції IgE, які до цього моменту, найбільш імовірно, не отримували належних активаційних сигналів [31].
Таким чином, протягом останніх років суттєво доповнилися знання щодо епідеміології, клінічної картини, ускладнень, критеріїв діагностики та раціональних підходів до лікування ізольованого дефіциту IgE у людей, про що мають бути ґрунтовно поінформовані лікарі різних спеціальностей.
Ізольований дефіцит IgE — дуже поширена в популяції первинна імунодефіцитна хвороба, яка має стати рутинною практикою клінічного імунолога. За частотою її випереджає лише вибірковий дефіцит IgD, що уразив щонайменше 6–8 % людей сучасної популяції. Відомі й зворотні феномени — так звані гіпер-IgE- та гіпер-IgD-синдроми, вивчення яких дозволило краще зрозуміти важливе біологічне призначення молекул IgE та IgD в організмі людини. Клінічна картина ізольованого дефіциту IgE зумовлена браком біологічних функцій цього класу імуноглобулінів і включає різнорідні синдроми, які формуються в різні періоди онтогенезу й поєднуються між собою у варіабельній манері. Йдеться про інфекційні, алергічні, псевдоалергічні, автоімунні, імунозапальні, онкологічні та деякі додаткові прояви, що можуть погіршити якість життя або навіть зумовити передчасну смерть пацієнта [29]. Характеризуючись високою частотою й гетерогенною клінічною картиною, ізольований дефіцит IgE несприятливо впливає на стан здоров’я сучасної людини, зумовлюючи великі прямі й непрямі затрати суспільства і держави, пов’язані з імунозалежною патологією. Тільки вдумливе, прискіпливе ставлення до проблеми своєчасної діагностики й раціонального лікування цієї імунодефіцитної хвороби дозволить досягнути істотного прогресу в подоланні її виражених негативних наслідків.
Конфлікт інтересів. Не заявлений.
Список литературы
1. Galkin O, Myhal’chuk M, Kazmirchuk VJe, Gurzhenko Ju. Development and comparison of various modifications of enzymoimmunoassay to determine specific IgE antibodies. Visnyk Kyi’vs’kogo nacional’nogo universytetu imeni Tarasa Shevchenka. 2014;1(66):15-21. (in Ukrainian).
2. Kazmirchuk VJe, Mal’cev DV, Caryk VV. Clinical picture, diagnosis and treatment of isolated IgE; literature review and clinical cases description. Klinichna imunologija, alergologija, infektologija. 2013;6-7(65-66):12-21. (in Ukrainian).
3. Kazmirchuk VJe, Mal’cev DV, Caryk VV, Cvejuk V. New view on events sequences in humoral immune response by Collins and Jackson concept. Imunologija ta alergologija. 2015;3-4:50-59. (in Russian).
4. Mal’cev DV. Association of autoimmune demyelinating disease of the nervous system with humoral immunodeficiency; new key to understanding effectiveness of immunotherapy. Mezhdunarodnyi Nevrologicheskii Zhurnal. 2014;2(64):27-32. (in Russian).
5. Mal’cev DV, Fedirko VO, Lisjanyj MI, Vasyl’jeva IG. Refractory atypical neuralgia associated with reactivated herpes viral infection: pathogenic association and effectiveness of combined antiviral treatment. Imunologija ta alergologija. 2014;3:62-77. (in Ukrainian).
6. Caryk VV, Kazmirchuk VJe. The characteristic of humoral immunity in chronic refractory rhinosinusitis. Imunologija ta alergologija. 2014;3:84-92. (in Ukrainian).
7. Ammann AJ, Hong R, Good RA. Healthy IgE-deficient person. N Engl J Med. 1970;283(10):542. doi: 10.1056/NEJM197009032831019
8. Bayry J, Fournier EM, Maddur MS, et al. Intravenous immunoglobulin induces proliferation and immunoglobulin synthesis from B cells of patients with common variable immunodeficiency: a mechanism underlying the beneficial effect of IVIg in primary immunodeficiencies. J Autoimmun. 2011;36(1):9-15. doi: 10.1016/j.jaut.2010.09.006
9. Bertoli LF, Pappas DG, Barton JC, Barton JC. Serum immunoglobulins in 28 adults with autoimmune sensorineural hearing loss: increased prevalence of subnormal immunoglobulin G1 and immunoglobulin G3. BMC Immunol. 2014;15:43. doi: 10.1186/s12865-014-0043-2
10. Cain VA, Ammann AJ, Hong R, et al. IgE deficiency associated with chronic sinopulmonary infection. J Chin Invest. 1969;48:12A.
11. Famuyiwa F, Rubinstein I. Chronic Sinopulmonary Inflammatory Diseases in Adults with Undetectable Serum IgE in Inner-City Chicago: A Preliminary Observation. Lung. 2012;190(3):291-4. doi: 10.1007/s00408-012-9375-y
12. Fulcher DA, Katelaris CH. Anaphylactoid reactions to local anaesthetics despite IgE deficiency: a case report. Asian Pac J Allergy Immunol. 1990;8(2):133-136.
13. García Pavón Osorio S, López Tiro JJ, Gómez Vera J. IgE deficiency: a forgotten disease? Rev Alerg Mex. 2009;56(6):192-197. (in Spanish).
14. Kashiwakura J, Kawakami Y, Yuki K, et al. Polyclonal IgE induce mast cell survival and cytokine production. Allergol Int. 2009;58(3):411-9. doi: 10.2332/allergolint.08-OA-0080
15. Kazmirchuk VE, Maltsev DV. Isolated IgE deficiency: clinical picture, diagnosis and treatment. In: VI World Asthma, Allergy and COPD Forum. 2013 April 27-30; London, UK. 21 p.
16. Kazmirchuk VY, Tsaryk VV, Sydorenko OI, et al. Frequency of immunoglobulin E deficiency among patients with immunodependent disorders. Likarska Sprava. 2014;7-8:3-9. (in Ukrainian).
17. Lam V, Kalesnikoff J, Lee CW, et al. IgE alone stimulates mast cell adhesion to fibronectin via pathways similar to those used by IgE + antigen but distinct from those used by Steel factor. Blood. 2003;102(4):1405. doi: 10.1182/blood-2002-10-3176
18. Magen E, Mishal J, Vardy D. Selective IgE deficiency and cardiovascular diseases. Allergy Asthma Proc. 2015;36(3):225-9. doi: 10.2500/aap.2015.36.3825
19. Magen E, Schlesinger M, Ben-Zion I, Vardy D. Helicobacter pylori infection in patients with selective immunoglobulin E deficiency. World J Gastroenterol. 2015;21(1):240-5. doi: 10.3748%2Fwjg.v21.i1.240
20. Magen E, Schlesinger M, David M. Selective IgE deficiency, immune dysregulation, and autoimmunity. Allergy Asthma Proc. 2014;35(2):e27-33. doi: 10.2500/aap.2014.35.3734
21. Maltsev DV, Kazmirchuk VE. Herpesvirus neuroinfections in immunocompromised patients: diagnosis and preventive treatment. In: Allergy, asthma and immunopathology: from basic science to clinical management. 2013 April 27-30. London.
22. Maltsev DV, Tsaryk VV, Kazmirchuk VE. Frequency of immunoglobulin E deficiency among immunologically evaluable patients. In: VII World Congress on immunopathology, respiratory allergy and asthma, 2013 October 12-15. Dubai, UAE.
23. McVicker S, Karim MY. IgE deficiency may indicate underlying hypogammaglobulinaemia? J Clin Pathol. 2014;67(9):832-3. doi: 10.1136/jclinpath-2014-202461
24. Pate MB, Smith JK, Chi DS, Krishnaswamy G. Regulation and dysregulation of immunoglobulin E: a molecular and clinical perspective. Clin Mol Allergy. 2010;8:3. doi: 10.1186/1476-7961-8-3
25. Polmar SH, Waldmann TA, Terry WD. IgE in immunodeficiency. Am J Pathol. 1972;69(3):499-512.
26. Riyaz N, Sasidharanpillai S, Rahima S, et al. Pyoderma gangrenosum in association with microscopic colitis, idiopathic hypereosinophilic syndrome, selective IgE deficiency and diabetes mellitus. Clin Exp Dermatol. 2015; Feb 22 40(6):629. doi: org/10.1111/ced.12618
27. Roa S, Isidoro-Garcia M, Davila I, et al. Molecular analysis of activation-induced cytidine deaminase gene in immunoglobulin-E deficient patients. Clin Dev Immunol. 2008;2008:146715. doi: 10.1155/2008/146715
28. Schoettler JJ, Schleissner LA, Heiner DC. Familial IgE deficiency associated with sinopulmonary disease. Chest. 1989;96(3):516-21. doi: 10.1378/chest.96.3.516
29. Smith JK, Krishnaswamy GH, Dykes R, et al. Clinical manifestations of IgE hypogammaglobulinemia. Ann Allergy Asthma Immunol. 1997;78(3):313-8. doi: 10.1016/S1081-1206(10)63188-2
30. Tamburin S, Borg K, Caro XJ, et al. Immunoglobulin G for the treatment of chronic pain: report of an expert workshop. Pain Med. 2014;15(7):1072-82. doi: 10.1111/pme.12319
31. Tsaryk VV. Treatment of refractory rhinosinusitis in patients with IgE deficiency. Likuval’na Sprava. 2014;12:3-10. (in Ukrainian).
32. Vengen IT, Madsen HO, Garred P, et al. Mannose-binding lectin deficiency is associated with myocardial infarction: the HUNT2 study in Norway. PLoS One. 2012;7(7):e42113. doi: 10.1371/journal.pone.0042113

/9-1.gif)
/10-1.gif)
/10-2.gif)
/12-1.gif)