Болезнь Олбрайта (БО, наследственная остеодистрофия, псевдогипопаратиреоз) — редкое наследственное полисистемное заболевание, характеризующееся невосприимчивостью клеток-мишеней к паратгормону [1, 2, 5]. Его истинная частота неизвестна, однако считается, что БО относится к числу орфанных болезней, так как в мировой литературе к концу XX века было описано только около 300 случаев [2, 3]. По механизму развития БО напоминает гипопаратиреоз, в основе которого лежит первичный дефицит паратиреоидного гормона, обусловленный снижением функциональной активности паращитовидных желез. При БО, напротив, клинические проявления вызываются нарушением чувствительности тканей-мишеней к действию паратгормона при его достаточном уровне секреции. Впервые эта патология описана в 1942 г. Ф. Олбрайтом (F. Albright) и в последующем получила эпонимический термин [8]. Заболевание обычно проявляется у детей и подростков [1, 3].
Этиология БО неизвестна, хотя считается, что
это генетически гетерогенное заболевание. Однако данные о типе ее наследственной передачи в литературе противоречивы. При БО описываются X-сцепленный, аутосомно-доминантный и аутосомно-рецессивный типы [1, 2, 7, 11]. В большинстве случаев развитие БО связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1. Наиболее часто наблюдается интерстициальная делеция длинного плеча второй хромосомы в локусе 2q37 [10]. Изучение родо–словных показывает, что число женщин с БО в 2 раза превышает количество мужчин, так как установлено, что БО не передается от отца к сыновьям [8]. В основе патогенеза БО лежит генетически обусловленная резистентность почек и костей к действию паратгормона в результате дефекта комплекса «специфический циторецептор — паратгормон — аденилатциклаза», что нарушает процесс образования в почках циклического 3’,5’-АМФ, являющегося внутриклеточным посредником действия паратгормона на метаболические процессы [7].
В зависимости от биохимического уровня поражения выделяют несколько типов БО [2, 3, 6].
1-й А тип: дефектен сам циторецептор, связывающий паратгормон. Имеет аутосомно-доминантный тип наследования. При данном типе нередко наблюдается одновременное вовлечение многих эндокринных желез: щитовидной и поджелудочной желез, гонад.
1-й В тип: отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном слое клеточной мембраны, который функционально связывает рецептор с аденилатциклазой. Также имеет аутосомно-доминантный тип наследования, однако не исключен сцепленный с Х-хромосомой тип. В отличие от 1-го А типа для данного типа нехарактерно поражение эндокринных желез.
2-й тип обусловлен ферментативной недостаточностью аденилатциклазы. Паратгормон при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на паратгормон в виде увеличения экскреции циклического аденозинмонофосфата (цАМФ). Однако внутриклеточная нечувствительность к цАМФ не позволяет в полной мере реализоваться действию паратгормона. Отличие данного типа от остальных форм БО заключается в том, что сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции цАМФ с мочой.
Некоторые авторы [6, 10] выделяют БО, которая характеризуется отсутствием гипокальциемии, гиперфосфатемии, судорог и остеомаляции, и относят ее к 1-му С типу. Однако вероятно, что этот вариант является одним из клинических фенотипов 1-го А типа [11].
Клиническая картина БО отличается мультисистемным и мультиорганным поражением, при котором соматические симптомы комбинируются с признаками поражения нервной системы [1–3]. У пациентов отмечаются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей, резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), «лунообразное» лицо [10]. Вследствие нарушений минерального обмена иногда наблюдаются экзостозы и аплазия зубов; компенсаторная гиперплазия паращитовидных желез (наличие в них аденом не характерно); кальцификация мягких тканей, подкожные кальцификаты; может отмечаться остеопороз; поражение глаз (лентикулярная катаракта); кальцинаты в подкожной клетчатке с тенденцией к их изъязвлению, что имитирует оссифицирующий миозит. В части случаев при БО наблюдается одновременное вовлечение эндокринных желез, что проявляется гипотиреозом, гипогонадизмом, сахарным диабетом, усугубляет течение БО и придает его клиническим проявлениям выраженный полиморфизм [1, 6].
Неврологические проявления считаются ведущими нарушениями при БО, так как обусловлены симметричной интрацеребральной кальцификацией коры полушарий, базальных ганглиев и зубчатых ядер мозжечка в связи с отложением солей кальция и железа в стенках мелких артерий и артериол, а также в веществе головного мозга [1, 2, 4]. Выделяют несколько ведущих клинических синдромов, которые у пациентов с БО встречаются в различных комбинациях и разной степени выраженности. К ним относят: 1) подкорковую деменцию; 2) локальные судороги и/или тетанические спазмы; 3) экстрапирамидные нарушения: гиперкинезы (хорея, тремор, дистония, атетоз, орофациальная дискинезия) или паркинсонизм; 4) мозжечковые симптомы; 5) нарушение речи; 6) преходящие или стойкие пирамидные знаки; 7) эпилептические приступы; 8) боль в дистальных отделах конечностей; 9) положительные симптомы Хвостека и Труссо [1, 2, 5, 11, 12].
К основным диагностическим критериям БО относят [3, 4, 6, 11]:
— низкий рост;
— круглое лицо;
— задержку нервно-психического развития;
— скелетные аномалии;
— низкое содержание кальция в сыворотке крови;
— высокий уровень паратиреоидного гормона в крови;
— снижение экскреции с мочой фосфатов и цАМФ.
Диагностика БО помимо характерной клинической картины основывается на лабораторных данных. Дифференциально-диагностическим тестом может быть характер почечной экскреции цАМФ в ответ на введение паратгормона: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие — при типе 1 [7, 10]. Диагноз БО подтверждается обнаружением сниженного уровня гуаниннуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой [7, 9]. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень паратгормона повышен, однако при 1C-типе его уровень может быть в норме [2, 6]. При рентгенологическом исследовании костной системы у части пациентов с БО обнаруживается укорочение пястных и плюсневых костей, нередко — генерализованная деминерализация (остеопороз), значительное утолщение костей свода черепа [1–3].
Дифференциальная диагностика БО проводится с истинным гипопаратиреозом; синдромами Аарскога, Барде — Бидля; болезнью Фара; Галлервордена — Шпатца; Шерешевского — Тернера; Вильсона — Коновалова; Гентингтона; Бассена — Корнцвейга; паразитарным поражением нервной системы (токсоплазмоз, эхинококкоз, цистицеркоз); туберозным склерозом Бурневилля [2, 5].
Лечение БО заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови [2, 3]. Большое значение имеет терапия витамином D. Начальную дозу рассчитывают из 2000 МЕ/кг массы тела в сутки, но не более 100 000 МЕ в сутки. Во избежание передозировки препаратов витамина D необходим контроль за концентрацией кальция в крови каждые 3–7 дней в течение первых двух недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца [2]. Можно применять кальцитрин, дигидротахистерол, оксидевит, а также другие препараты активных форм витамина D. Важное значение придают диете с ограничением фосфора, что помогает нормализовать концентрацию кальция в крови и устранить симптомы вторичного гиперпаратиреоза [1, 12]. При недостаточности других желез внутренней секреции проводят заместительную коррекцию соответствующими гормонами. При неврологических нарушениях используют симптоматическую терапию: противосудорожные препараты; антиоксиданты, мозговые метаболиты; препараты леводопы; клоназепам [3, 11].
Прогноз при БО точно не определен, однако можно предположить, что своевременная диагностика и рациональная терапия этой патологии позволяют говорить о положительных прогнозах на жизнь и возможности контроля за течением заболевания [1–3].
Ранее (в 2005 году) мы уже приводили семейный случай диагностики БО [1], однако исключительная редкость описания спорадических случаев этой патологии в русскоязычной литературе побудила нас привести еще одно собственное клиническое наблюдение, при этом сопроводив его анализом современной литературы.
Пациент Г., 36 лет, холост, на момент обращения не работает, инвалид 3-й группы, образование среднее специальное. Жалобы при поступлении на насильственные движения в левых конечностях, больше в руке; онемение в левой руке, нарушение речи; плохой сон. Считает себя больным с июня 2001 г., когда появились периодические тонические сокращения мышц правой половины лица с присоединением насильственных вычурных движений в правых конечностях продолжительностью 10–15 секунд 5–6 раз в сутки, возникающие после провоцирующего фактора (перемена положения тела, физическая нагрузка, внезапное речевое обращение к пациенту). 26.11.2001 г. был госпитализирован в одно из неврологических отделений в г. Минске, где проведена компьютерная томография головного мозга и на основании заключения специалиста по лучевой диагностике установлен диагноз «болезнь Фара». При выписке была рекомендована постоянная противосудорожная терапия карбамазепином. К 2006 г. на фоне регулярного приема данного препарата двигательные пароксизмы регрессировали, однако появились и начали усиливаться когнитивные нарушения. За последние несколько лет появились насильственные движения в левых конечностях, нарастала выраженность когнитивных нарушений, присоединилось нарушение речи. В течение всего периода заболевания пациент неоднократно проходил лечение в различных неврологических отделениях г. Минска с диагнозом «болезнь Фара». Анамнез жизни: отмечается патология перинатального периода: осложненные роды у матери, у новорожденного выявлен врожденный порок сердца (недостаточность митрального клапана). Отмечался логоневроз с детства, начал говорить только с 4 лет. Семейно-наследственный анамнез достоверно не отягощен, однако мать также имеет низкий рост и множественные стигмы строения скелета. В 1996 г. оперирован по поводу врожденного косоглазия. С 2003 по 2006 г. проходил лечение у офтальмологов с диагнозами: нейроретинит ОS, кератопатия ОS, артифакия, незрелая, осложненная катаракта ОD; оперировали катаракту ОS (2005 г.).
При объективном осмотре: общее состояние удовлетворительное, выглядит моложе своих лет, телосложение нормостеническое, алиментарное ожирение. Костно-мышечная система: низкий рост, равновеликая длина пальцев стоп, размер черепа непропорционален скелету, короткие широкие фаланги пальцев. АД 120/80 мм рт.ст., границы сердца не расширены, тоны сердца ясные, ритмичные; дыхание везикулярное. Язык влажный, не обложен, живот мягкий, безболезненный, печень не увеличена, мочеиспускание, стул — без особенностей, симптом Пастернацкого отрицательный. Неврологический статус: в сознании, эйфоричен, снижена память, ослаблено внимание, элементы брадифрении. Глазодвигательных нарушений, нистагма нет. Лицо симметрично. Язык по средней линии. Речь умеренно дизартрична, доступна пониманию. Объем активных и пассивных движений в конечностях сохранен. Несколько повышен тонус в левых конечностях, по экстрапирамидному типу — больше в руке. Глубокие рефлексы равномерно оживлены, D = S. Силовых парезов не выявлено. Почти постоянный среднеамплитудный неритмичный хореоатетоидный гиперкинез в левых конечностях, больше выражен в руке. В позе Ромберга устойчив. Координаторные пробы выполняет удовлетворительно. Нейропсихологическое тестирование: MMSE — 26 баллов, FAB — 8 баллов (характерны для умеренного подкоркового типа когнитивного снижения).
Лабораторные исследования. Общеклинические анализы крови и мочи — без особенностей. Биохимический анализ крови: кальций — 1,32 ммоль/л (норма 2,2–2,65 ммоль/л); фосфор — 2,03 ммоль/л (норма 0,81–1,45 ммоль/л). Гормональное исследование крови: АТ-ТПО — 63,06 мМЕ/мл (норма 0–34 мМЕ/мл),
Т3св. — 2,96 пм/л (норма 3,9–6,0 пм/л), Т4св. — 8,89 пм/л (норма 12–22 пм/л), остеокальцин — 43,55 пг/мл (норма 24–70 пм/л), паратгормон — 348,3 пг/мл (норма 15–65 пм/л), ТСГ — 5,78 мМЕ/мл (норма 0,5–4,67 мМЕ/мл), кальций ионизированный — 0,32 ммоль/л (норма 1,0–1,2 ммоль/л). Эти результаты указывали на гипофункцию щитовидной железы с аутоиммунным компонентом. Содержание паратгормона в несколько раз превышало норму. Уровень кальция в крови (общий и ионизированный) был ниже нормальных величин, а фосфора — напротив, повышен. Данный дисбаланс является патогномоничным для БО.
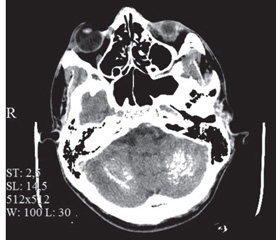
Инструментальные исследования. Сцинтиграфия паращитовидных желез: данных в пользу гиперфункциональной гормонально активной аденомы паращитовидных желез не выявлено. Остеоденситометрия поясничного отдела позвоночника и шеек бедренных костей: незначительная остеопения шейки бедренной кости слева. УЗИ щитовидной железы: диффузная гиперплазия. Рентгенография коленных суставов: рентген-признаки гонартроза 1–2-й ст., множественные мелкие очаговые тени (обызвествления), местами сливного характера, в мягких тканях параартикулярно. ЭЭГ: умеренные изменения ЭЭГ дизритмичного характера с признаками дисфункции срединно-стволовых структур головного мозга (S > D), патологических форм активности, очаговых нарушений нет. КТ головного мозга: линейные участки обызвествления в базальных ганглиях; пятнистые участки обызвествления белого вещества в лобных, теменных, затылочных и височных долях головного мозга (рис. 1), обоих полушариях мозжечка и проекции ствола (рис. 2), утолщены кости свода черепа (рис. 1).
/128-1.jpg )
Осмотры специалистов. уролог: без острой урологической патологии; эндокринолог: рекомендовано исследование кальция общего и ионизированного, уровня паратгормона; психиатр: когнитивно-мнестически снижен, эмоциональная лабильность в структуре органической патологии центральной нервной системы; логопед: дизартрия.
Учитывая характер жалоб, данные анамнеза, соматического, неврологического статуса, результаты лабораторных и инструментальных методов исследования, пациенту был установлен диагноз: болезнь Олбрайта с умеренным хореоатетоидным гиперкинезом преимущественно в левой руке, когнитивными нарушениями подкоркового типа, псевдогипопаратиреозом. Пациенту назначены витамин D, препараты кальция, акатинол мемантин, топирамат, строгая диета. При осмотре в дальнейшем отмечена некоторая положительная динамика в отношении выраженности хореоатетоидного гиперкинеза.
Мы проводили дифференциальную диагностику со всеми заболеваниями, упомянутыми выше, которые могут сопровождаться схожими клиническими и рентгенологическими данными. Наибольшую сложность вызвала дифференциальная диагностика с болезнью Фара, по поводу которой пациент наблюдался в течение длительного времени. В табл. 1 представлены отличия между этими заболеваниями, анализ которых позволил нам остановиться на диагнозе БО.
/128-3.jpg )
Из таблицы следует, что в пользу БО указывали дизрафические признаки пациента (низкий рост, равновеликая длина пальцев стоп, размер черепа непропорционален скелету, короткие широкие фаланги пальцев), уровень паратгормона, значительно превышающий нормальные показатели; гипофункция щитовидной железы с аутоиммунным компонентом; начальные стадии остеопороза; превалирование в неврологическом статусе гиперкинетического синдрома; на компьютерной томографии головного мозга, помимо стриопаллидозубчатого кальциноза, отмечался также гиперостоз свода черепа (толщина костей черепа в среднем на 2,5 мм превышала средние величины); проба с паратгормоном не привела к каким-либо значительным изменениям уровня кальция в сыворотке крови.
Интерес к данному клиническому случаю обусловлен исключительной редкостью заболевания, сложностью дифференциальной диагностики и полиморфизмом его клинических проявлений. Несмотря на распространенность поражения вещества головного мозга по результатам нейровизуализации, в неврологическом статусе у пациента отсутствовали мозжечковые и пирамидные симптомы. Клинический диагноз в нашем случае требует дополнительной ДНК-диагностики для его окончательной верификации и оценки риска появления БО у потомства.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.

/128-2.jpg )