Роль цитокинов в течении пневмонии, вызванной Staphylococcus aureus
Развитие воспалительного ответа при пневмонии, вызванной Staphylococcus aureus, обусловлено действием про- и противовоспалительных цитокинов, хемокинов, интерферонов I, II и III типа, продуцируемых клетками иммунной системы в ответ на инфицирование организма. Уровень активности большинства данных молекулярных компонентов механизмов противоинфекционной защиты предопределяет предрасположенность или резистентность к развитию стафилококковой инфекции и характер течения заболевания. Основные гены, продукты которых участвуют в защите от бактерий Staphylococcus aureus, представлены в табл. 1.
Провоспалительные цитокины Интерлейкины семейства 1
IL-1β
Во время стафилококковой инфекции интерлейкин 1β (interleukin-1β — IL-1β) в основном способствует привлечению нейтрофилов в очаг поражения [19, 48]. Интерлейкин IL-1β, взаимодействуя с рецептором IL-1R многочисленных типов иммуноцитов, активирует внутриклеточный MyD88/IRAK/NF-kB сигнальный путь [55, 78, 113] и индуцирует продукцию хемокинов С-Х-С: CXCL1, CXCL2, CXCL8, которые способствуют рекрутированию нейтрофилов в пневмонический очаг [13, 27].
IL-16
Интерлейкин-16 (lymphocyte chemoattractant factor — LCF) был впервые описан в качестве хемокинового фактора для Т-клеток в 1982 году. В настоящее время продемонстрировано, что IL-16 экспрессирует иммунные (моноциты, эозинофилы, CD4+Т- и CD8+Т-, тучные, дендритные клетки) и неиммунные клетки. Данный интерлейкин функционирует как провоспалительный цитокин, который вызывает два основных эффекта: способствует рекрутированию CD4+Т-лимфоцитов, преимущественно Th1-клеток, и ингибирует Тh2-клетки. Также IL-16 индуцирует хемотаксис эозинофилов и дендритных клеток [3, 63].
Протеин SpA бактерий метициллин-резистентного штамма Staphylococcus aureus (methicillin-resistant Staphylococcus aureus — MRSA), взаимодействуя с TNFR1 и рецептором эпидермального фактора роста (epidermal growth factor receptor — EGFR) [72], индуцирует продукцию IL-16 не только моноцитарными, но и эпителиальными и эндотелиальными клетками. В отличие от NF-kB-зависимых цитокинов индукция синтеза IL-16 непосредственно связана с TNF-α-ассоциированным каскадом. Представляет интерес тот факт, что самостоятельно LPS и TNF-α не стимулируют продукцию –IL-16, в то время как SpA MRSA является мощным триггером синтеза IL-16 для большинства типов IL-16-продуцирующих клеток. Секреция IL-16 в респираторном тракте инфицированных мышей ассоциирована с возбуждением не только TNFR1, но и EGFR. Ограничение участия IL-16 не снижает активность рекрутирования фагоцитов, но приводит к снижению продукции хемокина KC/CXCL1, рекрутирующего нейтрофилы в очаг поражения легких [1]. Danielle Ahn и соавт. [1] считают, что результаты исследований стафилококковой инфекции у нокаутных мышей Il16-/- не позволяют в полной мере оценить роль IL-16 в патогенезе пневмонии, вызванной MRSA.
IL-33
Hui Yin и соавт. [125] показали, что пептидогликаны (peptidoglycan — PGN) и липотейхоевые кислоты (lipoteichoic acid — LTA) бактерий Staphylococcus aureus могут стимулировать продукцию IL-33 макрофагами через возбуждение TLR2-МАРК-AKT-STAT3 сигнального пути. В отличие от других представителей интерлейкинового семейства 1 (IL-1 и IL-18), которые участвуют в Th1-ответе иммунной системы, IL-33 преимущественно активирует Th2-ассоциированную иммунную реакцию [32, 114], индуцирует созревание и дегрануляцию эозинофилов и тучных клеток [52].
Введение экзогенного IL-33 ингибирует колонизацию MRSA [125]. Fang Lana и соавт. [57] показали, что предварительное введение экзогенного –IL-33 способствует повышению активности клиренса бактерий золотистого стафилококка у мышей. Авторы считают, что защитный эффект IL-33 связан с индукцией экспрессии хемокинового рецептора CXCR2 и усилением инфильтрации нейтрофилами очага поражения, а также повышением активности фагоцитоза бактерий Staphylococcus aureus нейтрофилами. С другой стороны, IL-33 взаимодействует с ST2-рецептором, который широко представлен на мембране CD4+Foxp3+Treg клеток легочной ткани. При взаимодействии IL-33 с ST2-рецептором у Treg-клеток повышается экспрессия фактора транскрипции GATA3 (канонического фактора Th2-ответа), что обусловливает потерю способности Treg-клеток ингибировать эффекторные Т-лимфоциты. Введение в респираторный тракт IL-33 совместно с антигеном сопровождается нарушением предварительно сформированной иммунологической толерантности в ткани легкого [18].
В последнее время установлено, что IL-33 является не только провоспалительным цитокином, но и представляет собой ключевой медиатор репарации слизистых оболочек и восстановления эпителия [74, 128].
Таким образом, IL-33 ингибирует колонизацию MRSA, способствует бактериальному клиренсу во время пневмонии, вызванной Staphylococcus aureus, и ускоряет репарацию пораженных тканей.
Интерлейкины семейства 6
IL-6
Во время стафилококковой инфекции продукция IL-6 как ключевого провоспалительного цитокина обусловлена активацией различных внутриклеточных сигнальных путей многочисленных типов клеток респираторного тракта [98]. Установлено, что возбуждение LTA и PGN Staphylococcus aureus рецептора TLR2 через MyD88/NF-kB сигнальный путь приводит к индукции синтеза IL-6 [60]. Взаимодействие SpA золотистого стафилококка с TNFR1 вызывает активацию фактора транскрипции NF-kB, а с EGFR — AP-1. Данные факторы транскрипции транслоцируются в ядро и индуцируют экспрессию IL-6 [81].
IL-6, взаимодействуя со своим рецептором и за счет активации JAK/STAT-, RAS/MAPK-, PI3K/AKT-ассоциированных сигнальных путей, индуцирует синтез антител активированными В-клетками, в сочетании с TGF-β способствует дифференцировке наивных CD4+Т-клеток преимущественно в Th17-клетки и ингибирует TGF-β-индуцированную дифференцировку Treg-клеток. Также IL-6 индуцирует выработку острофазовых белков (C-реактивного белка, SAA, фибриногена, гепцидина) гепатоцитами [44, 66, 117]. Плейотропный IL-6 является важнейшим активатором STAT3-ассоциированного сигнального пути, одним из основных регуляторов дифференцировки Th17-клеток [14] и триггером экспрессии IL-22 [61].
Интерлейкин 6 в контексте реакции врожденной иммунной системы на воздействие инфекционного агента в ранний период заболевания поддерживает начальную волну привлечения нейтрофилов в очаг поражения, в последующем способствует переключению на рекрутирование моноцитов, а затем активирует апоптоз нейтрофилов, что обусловливает подавление активности воспаления [107].
Во время MRSA-инфекции в респираторном тракте IL-6 преимущественно продуцируется макрофагами. Секретируемый IL-6, взаимодействуя с рецепторами клеток легочного эпителия, возбуждает STAT3-ассоциированный сигнальный путь и индуцирует продукцию антимикробного протеина Reg3γ (regenerating islet-derived 3 gamma), который оказывает бактериостатическое и бактерицидное действие на MRSA [21].
У мышей дикого типа (Il6+/+) LTA и PGN Staphylococcus aureus индуцируют развитие острого дозозависимого воспаления легких. Индукция высокими дозами LTA Staphylococcus aureus характеризуется нейтрофильной инфильтрацией очага поражения и повышением уровня концентрации IL-6, TNF-α,
KC/CXCL1 в бронхоальвеолярной жидкости. В то же время при воспалении легких, вызванном низкими дозами LTA Staphylococcus aureus, IL-6 играет противовоспалительную роль. Интересно отметить, что при воспалении легких, вызванном PGN Staphylococcus aureus, вне зависимости от дозы IL-6 проявляет провоспалительные свойства: способствует рекрутированию полиморфноядерных лейкоцитов, формированию абсцессов легочной ткани. У нокаутных мышей Il6-/- активность воспалительной реакции легочной ткани не зависит от введенной дозы LTA, а характер воспаления отличается относительно низким уровнем рекрутирования нейтрофилов [60].
Продемонстрировано, что после инкубации с LTA у макрофагов Il6-/- наблюдается значительно более активная продукция TNF-α, чем у макрофагов Il6+/+ [60]. По всей вероятности, данная зависимость продукции TNF-α обусловлена способностью IL-6 ингибировать транскрипцию гена TNF-α [91].
Таким образом, действие IL-6 привносит существенный вклад в течение пневмонии, вызванной Staphylococcus aureus, усиливая активность воспаления в ранний период и подавляя воспалительный процесс в поздний период заболевания.
Интерлейкины семейства 12
IL-12
Во время заболеваний, вызванных Staphylococcus aureus, наблюдается повышение экспрессии IL-12, который вместе с IL-17 играет доминирующую роль в патогенезе стафилококковой инфекции [3, 79]. Quang-Tam Nguyen и соавт. [79] продемонстрировали наличие зависимости течения стафилококковой пневмонии от уровня продукции –IL-12. Установлено, что нокаутные мыши Il12p35-/-, не экспрессирующие IL-12p35, высокочувствительны к MRSA, а комбинированная терапия линезолидом с экзогенным –IL-12 MRSA-индуцированной пневмонии у данных мышей способствует их выживанию практически во всех случаях. Введение экзогенного IL-12 способствует усилению продукции IFN-γ NK-клетками (NK — natural killer), но не CD4+ или CD8+ Т-лимфоцитами. Применение экзогенного IL-12 способствует повышению уровня клиренса MRSA легочной ткани за счет активации NK-клеток и рекрутирования нейтрофилов. Уровень протективного влияния IL-12 зависит от активности –IL-12-индуцибельной продукции IFN-γ NK-клетками и коррелирует с экспрессией рецептора IFN-γ (IFNGR) у фагоцитов. Необходимо отметить, что терапия экзогенным IL-12 эффективна только при ее назначении до инфицирования Staphylococcus aureus и не эффективна при введении IL-12 через 4 часа после бактериального заражения. Учитывая, что у мышиных особей с обедненным представительством NK-клеток после введения –IL-12 не отмечается усиления бактериального клиренса легких, авторы считают, что именно NK-клетки играют ключевую роль в –IL-12-индуцированном саногенезе при пневмонии, вызванной MRSA. Возможно, применение рекомбинантных форм IL-12 потенциально может стать дополнительным методом лечения MRSA-индуцированных пневмоний, протекающих с высоким риском летального исхода.
В ранний период инфекционного процесса –IL-12 преимущественно активирует продукцию IFN-γ натуральными киллерами и Th1-клетками [130].
В то же время существуют данные, что –IL-12 способствует стафилококковой колонизации [106] и препятствует благоприятному течению Staphylococcus aureus-индуцированного абсцесса головного мозга [40], а назначение препаратов биологической терапии (в частности, устекинумаба), подавляющих активность IL-12, пациентам с неконтролируемым воспалением, вызванным Staphylococcus aureus, приводит к выздоровлению пациентов [49].
Интерлейкины семейства 17
IL-17А
Интерлейкин IL-17А представляет собой провоспалительный цитокин, который взаимодействует со своим рецептором, представленным на различных типах клеток, и индуцирует экспрессию цитокинов, хемокинов и металлопротеиназ. Нарушения Th17-ассоциированных сигнальных путей у человека, обусловленные генетическими причинами (например, гипер-IgE синдром), характеризуются высокой частотой встречаемости стафилококковой инфекции легких и кожи, что указывает на особую роль IL-17 в антистафилококковой защите [110].
Цитокиновый убиквитарный рецептор IL-17RA представлен на цитоплазматической мембране многочисленных иммунных и неиммунных клеток, включая эпителиоциты и фибробласты. Активация IL-17RA эпителиальных клеток респираторного тракта индуцирует продукцию группы провоспалительных цитокинов, хемокинов, способствуя инфильтрации эффекторными клетками пораженной ткани и развитию воспаления, синтезу молекул адгезии и антимикробных пептидов (АМП) [126]. Так, в результате действия IL-17A на эпителиальные клетки дыхательных путей усиливается продукция IL-6, IL-8/CXCL8, CXCL1, CXCL2, KC/СХС лиганда 1 (CXC ligand 1 — CXCL1) и GM-CSF, которые способствуют рекрутингу нейтрофилов в респираторный тракт; CCL20 и IL-19, привлекающих Th17- и Th2-клетки соответственно; CCL28, участвующего в вербовке IgE-секретирующих В-клеток. IL-17A повышает экспрессию ICAM-1 в эпителиальных клетках респираторного тракта. Установлено, что IL-17A индуцирует продукцию таких АМП, как CCL20, DEFB4, CRAMP, MUC5B/AC, S100A7, S100A8 и LCN2/24p3. Также IL-17A усиливает пролиферацию эпителиальных клеток респираторного тракта [64, 120].
Результаты экспериментальных исследований показали, что IL-17-дефицитные мыши более восприимчивы к бактериальным инфекционным агентам, в том числе и Staphylococcus aureus, чем мыши дикого типа [67, 77, 84].
Nathan K. Archer и соавт. [5] считают, что для предупреждения колонизации и деколонизации Staphylococcus aureus слизистой носовой полости необходим Th17-ассоциированный иммунный ответ. Представляет интерес тот факт, что у IL-23p19-дефицитных мышей не наблюдается нарушений бактериального клиренса Staphylococcus aureus в носовой полости. Отсутствие влияния IL-23 на бактериальный клиренс Staphylococcus aureus, по всей вероятности, свидетельствует о том, что продукция IL-17A при колонизации бактерий Staphylococcus aureus носит IL-23-независимый характер, что подчеркивает незначительность роли CD4+Th17-клеток в продукции IL-17A. Авторами установлено, что именно IL-17А играет ключевую роль в предупреждении колонизации бактериями Staphylococcus aureus слизистой носовой полости. Инфицирование ротоглотки бактериями Staphylococcus aureus сопровождается повышением содержания IL-17A, IL-1β, CXCL1 в секрете носовой полости. В отличие от мышей дикого типа IL-17A-дефицитные мыши не способны эффективно элиминировать бактерии золотистого стафилококка при инфицировании слизистой оболочки носовой полости экспериментальных животных. Нарушение элиминации бактерий золотистого стафилококка из носовой полости у –IL-17A-дефицитных мышей обусловлено снижением уровня рекрутирования нейтрофилов. Другим механизмом, IL-17A-зависимые нарушения которого могут способствовать колонизации Staphylococcus aureus, является продукция АМП: дефензинов и кателицидина (LL-37) [4].
У мышей с комбинированным дефицитом –IL-17A и IL-17F (Il17a-/-/Il17f- -/-) развиваются спонтанные стафилококковые абсцессы слизистых носовой и ротовой полости [47].
Anupa Kudva и соавт. [54] продемонстрировали особенности IL-17-опосредованного иммунного ответа при пневмонии, вызванной бактериями Staphylococcus aureus. Основным, но не единственным источником IL-17 в Staphylococcus aureus-инфицированной ткани легких являются IL-17-продуцирующие-γδТ-клетки (γδТ17-клетки), и истощение γδТ17-клеток приводит к снижению уровня продукции IL-17 в ранней фазе инфекционного процесса [20]. В то же время введение экзогенного IL-23 через 4–6 дней после перенесенного гриппа, который сопровождается ингибированием продукции IL-17A, приводит к повышению продукции последнего и уровня представительства нейтрофилов в ткани инфицированного Staphylococcus aureus легкого [54]. Таким образом, вероятно, в ранний период стафилококковой пневмонии основным источником IL-17A являются γδТ17-клетки, а в поздний период заболевания — CD4+Т17-клетки.
Стафилококковая пневмония у нокаутных мышей Il17r-/-, лишенных рецептора L-17R, протекает с низким уровнем клиренса бактерий Staphylococcus aureus из ткани легких [54]. Во время развития пневмонии продукция IL-17A у мышей дикого типа сопровождается активным рекрутированием нейтрофилов в очаг поражения легкого, а у нокаутных мышей Il17r-/- наблюдается достоверно более низкий уровень привлечения нейтрофилов в ткань легкого [124] на фоне сохраненного уровня продукции TNF-α в первые 24 часа после инфицирования бактериями Staphylococcus aureus [54].
Представляет интерес тот факт, что стафилококковая инфекция, развившаяся на фоне или после острой инфекции, вызванной вирусом гриппа А (influenza A virus — IAV), сопровождается низкой активностью рекрутирования нейтрофилов в очаг поражения легких [7, 109].
Таким образом, IL-17A препятствует колонизации бактерий Staphylococcus aureus и представляет собой высокозначимый фактор саногенеза пневмонии, вызванной Staphylococcus aureus. Подавление продукции IL-17A, наблюдаемое при гриппе, способствует развитию бактериального осложнения в виде стафилококковой пневмонии. Также показано, что инфекции, вызванные IAV, приводят к снижению экспрессии IL-1β и IL-23 и, как следствие, подавлению продукции IL-17 и IL-22. Введение экзогенных IL-1β или IL-23 сопровождается повышением уровня экспрессии IL-17 и IL-22 в респираторном тракте коинфицированных мышей и улучшением клиренса бактерий Staphylococcus aureus [54, 95].
Семейство IL-10
IL-22
Stefanie Gauguet и соавт. [34] продемонстрировали, что нейтрализация анти-IL-22 антителами цитокина IL-22 до инфицирования Staphylococcus aureus приводит к повышенной восприимчивости к стафилококковым бактериям, и, наоборот, введение экзогенного IL-22 мышам во время инфицирования золотистым стафилококком способствует повышению их резистентности к Staphylococcus aureus. Авторы считают, что введение рекомбинантного IL-22 представляет собой потенциально новый подход к лечению стафилококковой пневмонии. Большая часть саногенетических эффектов IL-22 связана с его способностью активировать воспалительную реакцию и поддерживать барьерную функцию эпителия респираторного тракта [56]. Сам по себе IL-22 не обладает прямым противовирусным или антибактериальным действием, но индуцирует продукцию АМП (β-дефензинов, Reg3γ), опосредуя раннюю защиту макроорганизма от бактериальных патогенов [129].
TNF-α
TNF-α является плейотропным цитокином, который оказывает влияние почти на каждую дифференцированную клетку, индуцируя широкий спектр различных клеточных реакций, в том числе активацию, пролиферацию, дифференцировку, выживание и гибель клетки [2].
Цитокин TNF-α в основном синтезируется такими иммуноцитами, как макрофаги, дендритные клетки, Т-, В-лимфоциты, NK-клетки, тучные клетки, и продуцируется в виде гомотримера (34 кДа). Данный цитокин может функционировать в виде мембранно-связанной или солютабной формы. Солютабная гомотримерная форма образуется в результате отщепления мембранно-связанной формы TNF-α от поверхности мембраны клетки при помощи металлопротеиназы, которая представляет собой TNFα-превращающий фермент (TNFα-converting enzyme — ТАСЕ или ADAM metallopeptidase domain 17 — ADAM17). Цитокин TNF-α реализует свое действие, связываясь с трансмембранными рецепторами I типа: TNFR1 и TNFR2, обладающими одинаковым аффинитетом к данному цитокину. Рецептор TNFR1 преимущественно расположен на мембране аппарата Гольджи, а TNFR2, как правило, локализован на цитоплазматической мембране клетки. Рецептор TNFR1 может быть активирован как трансмембранной, так и солютабной формой TNF-α. Лигирование TNFR1 первоначально приводит к образованию Complex I, состоящего из TRADD (TNFR1-associated death domain protein), TRAF2 (TNFR-associated factor 2), RIPK1 (receptor-interacting serine/threonine-protein kinase 1), cIAP1 (cellular inhibitor of apoptosis protein 1), cIAP2 и комплекса линейных убиквитиновых цепей (linear ubiquitin chain assembly Complex — LUBAC). Данный Complex I активирует ядерный фактор –NF-κB и митоген-активируемые протеинкиназы (mitogen-activated protein kinase — МАРК). Формирование Complex I индуцирует выживание и пролиферацию клеток, развитие воспаления, пироптоз и активацию иммунных механизмов защиты от патогенных микроорганизмов. Образование пироптосомы — Complex IIa, содержащего RIPK1, TRADD, FADD (Fas associated via death domain), прокаспазу 8 и cFLIPL/CFLAR, или Complex IIb, содержащего RIPK1, RIPK3, FADD прокаспазу 8, индуцирует апоптоз клетки, а Complex IIc (некросомы), состоящего из RIPK1, RIPK3, вызывает некроптоз и воспаление. Рецептор TNFR2 активируется трансмембранной формой TNF-α, вследствие чего он вербует TNFR-ассоциированный фактор 2 (TNFR-associated factor 2 — TRAF2), индуцируя образование Complex I. Рецептор TNFR2 в основном обеспечивает гомеостатическую биологическую активность, включая регенерацию тканей, пролиферацию и выживаемость клеток [50, 65, 93].
Бактерии Staphylococcus aureus индуцируют продукцию TNF-α. Экспериментально установлено, что в клетках человеческой моноцитарной линии (ТНР-1 клетках) после их взаимодействия со стафилококковым энтеротоксином B (SEB) достоверно повышается экспрессия TNF-α [127].
Необходимо отметить, что инфекция, вызванная золотистым стафилококком, приводит к специфической активации рецептора TNFR1, но не TNFR2 [6]. Продемонстрировано, что TNF-α играет ключевую роль в развитии местного воспалительного процесса, но активация его вызывает также неблагоприятные системные эффекты. Так, введение рекомбинантного TNF-α экспериментальным животным вызывает артериальную гипотензию, метаболический ацидоз, выраженную гемоконцентрацию, шок и летальный исход в течение нескольких минут или часов [119, 122]. Однако применение анти-TNF-α терапии (в частности, этанерцепта) при лечении больных с септической стафилококковой инфекцией не способствует процессу выздоровления [22]. При инфекциях респираторного тракта LPS-индуцированная продукция TNF-α вызывает активацию р38 митогенактивируемой протеинкиназы, сопровождаясь рекрутированием нейтрофилов в очаг поражения легких и развитием бронхоспазма. Представляет интерес, что TLR4-опосредованное воспаление легочной ткани зависит от уровня экспрессии CD14/MD2 и адаптерных молекул TIRAP и MyD88, в то время как TRIF, –IL-1R1 и IL-18R-ассоциированные сигнальные пути являются необязательными компонентами [118].
Во время стафилококковой инфекции с рецептором TNFR1 взаимодействует не только TNF-α, но и протеин SpA — один из основных внеклеточных факторов вирулентности бактерий Staphylococcus aureus. SpA-лигирование TNFR1 вызывает активацию фактора транскрипции NF-kB и последующее развитие нейтрофильно-макрофагального воспаления [72]. TNF-лигирование TNFR1 также активирует некроптоз и апоптоз. Также протеин SpA бактерий Staphylococcus aureus индуцирует мобилизацию рецептора TNFR1 к апикальной поверхности мембраны эпителиоцитов. Взаимодействие протеина SpA с TNFR1 иници–ирует продукцию хемокинов IL-8/CXCL8, CXCL10 эпителиальными клетками дыхательных путей и макрофагами. Продуцируемые хемокины обусловливают вербовку нейтрофилов в очаг поражения респираторного тракта, способствуя бактериальному клиренсу. Однако SpA бактерий Staphylococcus aureus во время системного инфекционного процесса в естественных условиях индуцирует раннее отщепление рецептора TNFR1 от мембраны клетки, которое предшествует высвобождению TNF-α. Результаты, полученные в экспериментах с использованием мутантных SpA-дефицитных бактерий Staphylococcus aureus и мышей Tnfr1-/-, убедительно свидетельствуют о том, что циркулирующие солютабные формы TNFR1, образование которых индуцировано золотистым стафилококком, нейтрализуют TNF-α и подавляют воспалительную реакцию в начальной фазе пневмонии [35]. SpA-индуцированное рекрутирование нейтрофилов в очаг поражения легких является решающим фактором, обеспечивающим эрадикацию бактериальных патогенов. Так, у мышей с нокаутом гена Tnfr1 наблюдается значительно менее выраженная нейтрофильная инфильтрация в пневмоническом очаге, чем у мышей дикого типа. Так как наличие TLR-ассоциированной адаптерной молекулы MyD88 является необязательным условием для развития стафилококковой пневмонии в естественных условиях, по мнению ряда авторов, TNFR1 является основным сенсором, активируя который бактерии Staphylococcus aureus вызывают развитие воспаления в респираторном тракте [36, 113].
Индуцированный бактериями Staphylococcus aureus TNF-α вызывает развитие воспаления. Во время стафилококковой инфекции цитокин TNF-α, кроме воспаления, может индуцировать апоптоз клеток. Интересно, что SEB-индуцированный апоптоз Т-клеток и эпителиальных клеток связан с Fas-опосредованными процессами, в то время как апоптоз ТНР-1 клеток практически не сопровождается повышением концентрации мРНК Fas и TRAIL [121, 127].
Интерфероны
Развитие пневмонии, вызванной Staphylococcus aureus, сопровождается возбуждением образ-распознающих рецепторов, ассоциированных с активацией генов интерферонов (interferon — IFN) [53, 87, 89].
Интерфероны I типа
Продемонстрировано, что уже через 2 часа после инфицирования бактериями Staphylococcus aureus наблюдается усиление продукции IFN I типа клетками респираторного тракта [70, 86, 90].
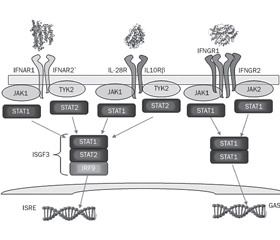
PAMP бактерий Staphylococcus aureus в респираторном тракте активируют продукцию IFN I типа, которые, взаимодействуя с собственными рецепторами, индуцируют факторы транскрипции: интерферон-регуляторные факторы (interferon regulatory factor — IRF), трансдукторы сигнала и активаторы транскрипции STAT1, STAT2 и STAT3 (signal transducer and activator of transcription — STAT). Взаимодействие IFN I типа, таких как IFN-β, с его гетеродимерным рецептором (IFNAR) через возбуждение рецепторассоциированных киназ — Janus киназы 1 (Janus kinase 1 — JAK1) и тирозинкиназы 2 (tyrosine kinase 2 — TYK2) — приводит к фосфорилированию факторов транскрипции STAT1, STAT2 и образованию димера STAT1/STAT2, который регулирует транскрипцию сотен интерферон-сенситивных генов, в том числе и гена протеина IRF-7, который индуцирует синтез IFN-α [71].
Индукция IFN-β в респираторном тракте Staphylococcus aureus осуществляется через возбуждение различных молекулярных путей, в том числе и через TLR9/IRF1 или NLRC2/RIP2/IRF5 пути [88].
В естественных условиях продукция IFN I типа в ответ на инфицирование Staphylococcus aureus способствует неблагоприятному течению заболевания. В частности, продемонстрировано, что при инфицировании бактериями Staphylococcus aureus у нокаутных мышей Ifnar-/-, с делецией гена рецептора интерферона Ifnar, значительно реже развивается пневмония, чем у мышей дикого типа. Стафилококковая пневмония у нокаутных мышей Ifnar-/- достоверно реже заканчивается летальным исходом. При инфицировании нелетальными дозами бактерий Staphylococcus aureus у мышей Ifnar-/- наблюдается достоверно более низкий уровень содержания TNF-α и IL-6 в бронхоальвеолярной жидкости, чем у мышей дикого типа при одинаковой бактериальной нагрузке в ткани легкого [70]. IFN I типа индуцируют продукцию TNF-α, который вносит существенный вклад в патогенез инфекционно-токсического шока [46]. Также у мышей Ifnar-/- наблюдается более высокий уровень Th17-ассоциированной реакции на протеогликан [29].
Установлено, что при стафилококковой пневмонии IFN I типа ингибируют продукцию дендритными клетками IL-23, который необходим для активации Th17-клеток. Стафилококковая инфекция у нокаутных мышей Ifnar-/- на фоне IAV-инфекции характеризуется более благоприятным течением, чем у мышей дикого типа. Повышенные концентрации IFN I типа могут препятствовать развитию Th17-ответа, обусловливая возникновение вторичных бактериальных инфекций, в том числе и вызванных бактериями Staphylococcus aureus. С другой стороны, снижение уровня секреции IFN I типа сопровождается увеличением активности Th17-ассоциированного звена воспаления [15, 54].
При лечении стафилококковой пневмонии необходимо учитывать тот факт, что высоковирулентные штаммы MRSA могут непосредственно индуцировать продукцию IFN I типа [70]. Kelly M. Shepardson и соавт. [111] считают, что IFN I типа на фоне гриппозной инфекции оказывают дифференцированное действие на развитие стафилококковой суперинфекции, и эффект этого действия определяется соотношением продукции IFN-α и IFN-β. Так, увеличение продукции IFN-β альвеолярными макрофагами (CD11c+CD11b−SiglecF+) в доклинической стадии IAV-инфекции (третьи сутки от момента инфицирования) коррелирует со сниженной вероятностью развития MRSA-суперинфекции, а увеличение продукции IFN-α моноцитами (CD11c+CD11b−SiglecF−Ly6C+) и нейтрофилами (CD11b+CD11c−Ly6C+) во время клинической стадии IAV-инфекции (седьмые сутки от момента инфицирования) сопряжено с высокой вероятностью развития MRSA-суперинфекции. Продукция IFN I типа Ly6G+-клетками в доклинической стадии IAV-инфекции сопровождается гибелью бактерий MRSA, а во время клинической стадии IAV-инфекции — способствует развитию суперинфекции за счет ингибирующего действия IFN I типа на активность Ly6G+-нейтрофилов. Согласно результатам, полученным авторами [101, 111], CD11c+-клетки, в большей степени альвеолярные макрофаги, в ответ на возбуждение IFNAR в доклиническом периоде гриппозной инфекции продуцируют IL-13, продукция которого достигает своего пика к третьим суткам от момента инфицирования. Интерлейкин IL-13 подавляет синтез IFN-γ и способствует повышению уровня резистентности организма к бактериям Staphylococcus aureus. Мутантные мыши с делецией гена Il13 (Il13-/-) отличаются повышенной продукцией IFN-γ и высокой склонностью к развитию пневмонии, вызванной MRSA, в доклинический период гриппозной инфекции. Установлено, что у мышей в течение первых трех суток гриппозной инфекции отмечается значительное снижение уровня сывороточного IL-13-связывающего протеина IL-13Rα2, в то время как в клиническом периоде (7-е сутки) наблюдается повышенная экспрессия IL-13Rα2, которая способствует развитию MRSA-суперинфекции. Назначение анти-IL-13Rα2 антител в ранний период приводит к увеличению бактериальной нагрузки, а в поздний период гриппозной инфекции — к уменьшению MRSA нагрузки в легочной ткани. Интересно отметить, что назначение экзогенного IL-13 мышам дикого типа на вторые — четвертые сутки гриппозной инфекции сопровождается увеличением уровня вирусной нагрузки [100].
Таким образом, IFN I типа предопределяют развитие стафилококковой инфекции на фоне острых респираторных вирусных заболеваний. Продукция IFN-β CD11c+- и Ly6+-клетками в первые трое суток (в доклиническом периоде) гриппозной инфекции препятствует развитию стафилококковой инфекции, в то время как продукция IFN-α Ly6+-клетками в клиническом периоде гриппозной инфекции (после шестого дня от момента инфицирования вирусами гриппа) способствует развитию стафилококковой инфекции. Индуцированная секреция IFN I типа на 5-й день после инфицирования IAV способствует подавлению продукции хемоаттрактантов CXCL1 и CXCL2, что приводит к уменьшению представительства нейтрофилов в очаге поражения легких [102]. IFN I типа подавляют продукцию –IL-1β, IL-23, которые являются критическими иммунорегуляторными цитокинами для Th17-клеток [95]. Согласно представленным данным, IFN I типа после 6-х и 7-х суток от момента инфицирования вирусами гриппа способствуют развитию стафилококковой суперинфекции. По мнению Michelle E. Mulcahy и Rachel M. McLoughlin [76], развитие вторичной бактериальной инфекции на фоне вирусного поражения является более сложным процессом, чем просто использование бактериальными агентами вирус-индуцированной повышенной восприимчивости макроорганизма.
Интерферон II типа (IFN-γ)
Единственным представителем интерферонов II типа является IFN-γ. Интерферон IFN-γ продуцируется CD4+Th1-, CD8+ T-цитотоксическими лимфоцитами, NK-, NKT-, В-, антигенпрезентирующими (APC) клетками. В раннем периоде инфекционного процесса основную часть IFN-γ продуцируют NK-клетки и, возможно, профессиональные APC клетки, в то время как развитие адаптивного иммунного ответа связано с продукцией IFN-γ Т-лимфоцитами. Cинтез IFN-γ индуцируют TNF-α и IL-12, IL-18, секретируемые APC клетками. Первоначальная продукция IFN-γ обусловлена IL-12-зависимой активацией NK-клеток, а последующая продукция IFN-γ — комбинированным действием IL-12 и IL-18 на макрофаги, NK- и Т-клетки [25, 85, 108]. Основным эффектом действия IFN-γ является активация сложной сети IFN-индуцируемых генов, в основном участвующих в реакции врожденной иммунной системы [108]. Предполагается, что IFN-γ индуцирует посттранскрипционные и/или эпигенетические изменения, которые отвечают за TLR-ассоциированную воспалительную реакцию и классическую активацию макрофагов [12, 43]. Вполне вероятно, что рекогниция PAMP патогенов TLR инициирует раннюю продукцию IFN-γ, который в последующем усиливает ответ врожденной иммунной системы через индукцию TLR. С другой стороны, IFN-γ ингибирует генерацию микроРНК-3473b, которая подавляет активацию макрофагов, непосредственно влияя на экспрессию гена фосфатазы и гомолога тензина (phosphatase and tensin homolog — PTEN), участвующего в регуляции продукции IL-10 [123]. Интерферон II типа вызывает многочисленные эффекты у клеток иммунной системы (табл. 2).
/137-1.jpg )
Интерферон II типа IFN-γ активно участвует в патогенезе пневмонии, вызванной бактериями Staphylococcus aureus, способствуя генерации активных кислородсодержащих метаболитов (АКМ), продукции хемокинов, привлекающих эффекторные клетки. Взаимодействие IFN-γ с собственным рецептором активирует JAK1 и STAT1 сигнальные пути через 15–30 минут, возбуждая транскрипцию сенситивных генов [108].
Так, IFN-γ увеличивает активность НАДФН оксидазы фагоцитов, СOX1 тучных клеток, обусловливая индукцию высокого уровня генерации АКМ, вызывающих гибель бактерий золотистого стафилококка [37, 115]. В отличие от быстро реагирующих генов пик повышения транскрипции генов, участвующих в организации НАДФН оксидазы, в макрофагах человека наблюдается на 3–4-й день действия IFN-γ [16].
Бактерицидная активность макрофагов человека проявляется уже в первые 90 минут индуцирующего действия IFN-γ, что приводит к снижению бактериальной нагрузки при стафилококковой инфекции [37].
Интерферон IFN-γ увеличивает чувствительность макрофагов к TLR-лигандам и к действию цитокинов [42]. Mallary C. Greenlee-Wacker и William M. Nauseef [37] продемонстрировали, что IFN-γ усиливает продукцию IL-6 и IL-1β макрофагами только на фоне инфицирования Staphylococcus aureus. В ответ на действие бактерий Staphylococcus aureus макрофаги, индуцированные IFN-γ, демонстрируют 4-кратное увеличение продукции IL-6 по сравнению с неиндуцированными макрофагами.
Во время стафилококковой инфекции действие IFN-γ вызывает продукцию взаимодействующих с рецептором CXCR3 хемокинов: CXCL9, CXCL10 и CXCL11, которые участвуют в рекрутинге Th1-, Th17-клеток и полиморфноядерных лейкоцитов [26, 38]. Блокирование рецептора CXCR3 сопровождается снижением представительства CD4+Т-клеток в бронхоальвеолярной жидкости и выраженности воспалительной реакции в очаге поражения легких [69].
Существуют научные данные о том, что IFN-γ, как и IL-12, может содействовать стафилококковой колонизации [106].
Интерфероны III типа (IFN-λ)
Бактерии Staphylococcus aureus активируют продукцию интерферонов III типа, которые вносят свой вклад в развитие воспаления при стафилококковой пневмонии [23]. Несмотря на схожесть функционирования IFN I и III типов, ассоциированные с ними сигнальные системы характеризуются рядом существенных отличий. Во-первых, рецептор IFNAR экспрессируется практически всеми клетками организма, а рецептор IFN-λ — IL-28R расположен главным образом на эпителиальных клетках и нейтрофилах. Во-вторых, реакция транскриптома на воздействие IFN I типа отличается ранним и сильным, а на влияние IFN III типа (IFN-λ) — пролонгированным и слабым ответом [10, 68, 87]. В то время как все IFN I типа взаимодействуют с общим гетеродимерным рецептором — IFNAR (IFNAR1 и IFNAR2), IFN-λ связываются с IFNLR, который представляет собой уникальный гетеродимерный рецептор, состоящий из двух субъединиц: общей с рецепторным семейством IL-10R субъединицы IL10Rβ, и специфической для IFN-λ субъединицы IFNLR1 или IL28Rα [58]. Интерфероны III типа, взаимодействуя со своим рецептором, активируют факторы транскрипции IRF-1, IRF-3, IRF-7 и –NF-kB, которые изменяют экспрессию таргетных генов [33].
Эпителиальные клетки респираторного тракта и миелоидные клетки человека продуцируют IFN-λ в ответ на инфекционные агенты, особенно вирусные. В структуре объема продукции интерферонов респираторными эпителиальными клетками после инфицирования вирусом гриппа или другими респираторными вирусами IFN-λ занимают первое место [30, 94].
В настоящее время не уточнены стафилококковые молекулярные компоненты, выступающие триггерами продукции IFN-λ. Индукция IL-28R сигнального пути активирует экспрессию АМП и молекулярных компонентов клеточного цитоскелета, которые регулируют барьерную функцию слизистой оболочки респираторного тракта. Считают, что с продукцией IFN-λ ассоциирован риск колонизации дыхательных путей патогенными микроорганизмами, в том числе и MRSA, способными вызвать воспаление легких [58, 92].
Согласно данным Paul J. Planet и соавт. [92], в респираторном тракте во время острой инфекции IFN III типа преимущественно активируют –IL-28R/STAT1/SOCS1 сигнальный путь. Цитокины IFN-λ в клетках слизистой оболочки носовой полости, активируя фактор транскрипции STAT1 и его регулятор SOCS1, способствуют росту колоний патогенных бактерий. IFN-λ подавляют активность воспалительного процесса, нарушая привлечение нейтрофилов и ингибируя продукцию IL-1β [10].
Продукция IFN-λ, так же как и IFN-α, способствует неблагоприятному течению стафилококковой инфекции. Так, мыши с делецией гена рецептора Il28r характеризуются высокой степенью резистентности к бактериям Staphylococcus aureus. У данных мышей стафилококковая пневмония протекает с более высоким уровнем бактериального клиренса при более низкой продукции цитокинов (IL-1β, GM-CSF, KC/CXCL1) и сниженном профиле активности воспаления легочной ткани. Избыток продукции IFN-λ способствует поражению легочной ткани при пневмонии, вызванной Staphylococcus aureus, особенно, штаммом USA300 MRSA [24]. Повышение резистентности у мутантных мышей Il28r-/-к Staphylococcus aureus, вероятно, обусловлено увеличением IL-22-зависимой продукции АМП [75]. Установлено, что у нокаутных мышей Il28r-/- заметно увеличена конститутивная экспрессия IL-22. Возможно, что экспрессия –IL-22 является компенсаторной реакцией на дефицит IFN-λ, так как и сигналы IFN-λ, и IL-22 необходимы для фосфорилирования фактора транскрипции STAT1 [41].
Интерфероновые сигнальные пути и их роль в развитии стафилококковой инфекции на фоне острой респираторной вирусной инфекции представлены на рис. 1.
Противовоспалительные цитокины Семейство IL-10
IL-10
Thu A. Chau и соавт. [17] показали, что PGN бактериальной стенки Staphylococcus aureus, взаимодействуя с TLR2 (в сочетании с TLR1 или TLR6) АРС, через активацию канонической NF-kB-ассоциированной сигнальной цепи вызывают продукцию IL-10, который ингибирует воспалительный ответ, преимущественно за счет подавления активности Th1-, Th17-клеток [31]. Представляет интерес тот факт, что возбуждение TLR2-ассоциированных сигнальных путей носит дифференцированный лиганд-зависимый характер. Так, вирусные, но не бактериальные TLR2-лиганды индуцируют продукцию IFN I типа моноцитами [8], липопротеины вызывают секрецию провоспалительных цитокинов [116], а некоторые лиганды, в частности PGN, усиливают экспрессию IL-10 [59]. Vanessa Frodermann и соавт. [31] установили, что возбуждение PAMP Staphylococcus aureus рецептора TLR2 в APC может вызывать продукцию IL-10 или цитокинов, участвующих в активации Th1-, Th17-клеток. Провоспалительный и противовоспалительный эффект TLR2-индуцированной реакции обусловлен участием аксессуарных молекул во взаимодействии стафилококковых лигандов с TLR2. Так, провоспалительный PGN/TLR2-индуцированный ответ моноцитов человека ассоциирован с участием молекулы CD14, в то время как противовоспалительный PGN/TLR2-индуцированный ответ не требует участия аксессуарных молекул CD14, CD36. Последующая селективная активация PI3K/AKT внутриклеточного сигнального пути индуцирует продукцию первичными моноцитами именно –IL-10. Молекулы TLR1, TLR2 и TLR6 содержат PI3K-связывающие мотивы, но не имеют внутренней киназной активности, и поэтому для фосфорилирования PI3K требуется участие другой киназы. Одним из возможных кандидатов для выполнения дифференциального рекрутинга и активации PI3K считают адаптерную молекулу T-клеточной дифференциации MAL (myelin and lymphocyte protein, T cell differentiation protein) [104] и Src-тирозинкиназу LYN (LYN proto-oncogene, Src family tyrosine kinase) или протеинтирозинфосфатазу 1 (protein tyrosine phosphatase, non-receptor type 6 — PTPN6) [51]. Моноциты и макрофаги являются основными продуцентами IL-10 в ответ на инфицирование золотистым стафилококком респираторного тракта [82]. Моноциты и макрофаги в 4–20 раз больше продуцируют IL-10, чем дендритные клетки [31]. Моноцитарные дендритные клетки в ответ на активацию TLR2 продуцируют преимущественно IL-12 и IL-23 [31].
IL-10 реализует свое действие, взаимодействуя с рецептором IL-10R, состоящим из двух субъединиц — IL-10R1 и IL-10R2. Возбуждение IL-10R активирует сигнальные пути, обусловливающие формирование активного димера STAT3/STAT3. Димер STAT3/STAT3, транслоцируясь в ядро клетки, изменяет транскрипцию таргетных генов, продукты которых подавляют активность провоспалительных сигнальных путей. В частности, под влиянием –IL-10 индуцируется синтез интерактивного протеина TNIP3 (TNFAIP3 interacting protein 3), который подавляет активацию фактора транскрипции NF-kB; дуальной специфической фосфатазы (dual specificity phosphatase 11 — DUSP11), которая предотвращает активацию p38 MAPK; IBCL3 (B-cell CLL/lymphoma 3), который ингибирует синтез TNF-α; атипичного инибитора NF-kB IκBNS, подавляющего экспрессию гена IL-6 [103]. Известно, что IL-10 ингибирует экспрессию антигенов II класса главного комплекса гистосовместимости и костимулирующих молекул АРС, что приводит к снижению активации Т-клеток. В отличие от ингибирующего влияния на клеточный иммунитет на гуморальное звено IL-10 оказывает стимулирующее влияние: он активирует пролиферацию B-клеток и усиливает синтез иммуноглобулинов [73, 80, 83, 99].
Введение рекомбинантного IL-10 мышам со стафилококковой инфекцией предотвращает развитие инфекционно-токсического шока, индуцированного суперантигенами [9]. С другой стороны, высокая концентрация IL-10 в связи с генерацией толерогенной иммунологической среды способствует стафилококковой колонизации слизистых оболочек респираторного тракта [62]. Также установлено, что повышенная концентрация IL-10 и PGN в сыворотке крови у пациентов со стафилококковой инфекцией, протекающей с бактериемией, ассоциирована с высоким риском летального исхода [97].
Таким образом, в период реконвалесценции стафилококковой пневмонии IL-10 способствует снижению активности процесса воспаления, но если инфекционный процесс сопровождается бактериемией, высокий уровень продукции IL-10 может выступать в качестве танатогенного фактора.
Интерлейкины семейства 12
IL-27
Интерлейкин-27 (IL-27) представляет собой гетеродимерный цитокин, состоящий из EBI3 и р28 субъединиц, который взаимодействует со своим гетеродимерным рецептором и за счет активации факторов транскрипции STAT1 и STAT3 может вызывать как про-, так и противовоспалительные эффекты. Димеризация субъединиц gp130 и IL-27Ra рецептора IL-27 рекрутирует к интрадомену JAK1, JAK2 и TYK2, которые передают возбуждение МАРК- и STAT (STAT1 и STAT3)-ассоциированным сигнальным каскадам. Активация STAT1 связана с ингибированием факторов транскрипции: протеина 3, связывающего последовательность GATA (GATA binding protein 3 — GATA-3), ретиноид-связанного рецептора γt (RAR related orphan receptor γt — RORγt), и усилением экспрессии генов: специ–фического для T-клеток фактора транскрипции (T-BET — T-cell-specific T-box transcription factor), молекулы CD274 и IL-10. Активация фактора транскрипции STAT3 также приводит к экспрессии IL-10, а возбуждение МАРК-ассоциированного сигнального пути — к индукции IL-10 и IL-21 [45]. В соответствии с дуальной ролью IL-27 предотвращает повреждение ткани, вызванное чрезмерным воспалением [131]. Так, установлено, что IL-27, активируя фактор транскрипции STAT-1, приводит к блокаде экспрессии RORγt, что подавляет дифференцировку наивных T-лимфоцитов в Th17-клетки [28]. Кроме того, IL-27 является основным триггером, индуцирующим продукцию IL-10 CD8+Т-клетками [105].
Keven M. Robinson и соавт. [96] продемонстрировали, что у нокаутных мышей Il27rα-/-, лишенных рецептора к IL-27, стафилококковая пневмония протекает с более низким уровнем инфильтрации нейтрофилами и макрофагами легочной ткани в очаге инфекционного поражения. У нокаутных мышей Il27rα-/- при сочетанном инфекционном процессе, вызванном Staphylococcus aureus и IAV, наблюдается увеличение активности бактериального клиренса в легочной ткани, которое сопровождается высоким уровнем продукции IL-17A и IL-17F в комбинации с относительно низким уровнем продукции IL-10 по сравнению с мышами дикого типа. Согласно полученным данным, авторы считают, что во время IAV-инфекции действие IL-27, индуцирующее продукцию IL-10 и подавляющее синтез IL-17, предрасполагает к развитию пневмонии, ассоциированной с золотистым стафилококком. В то же время IL-27-индуцированный IL-10 предупреждает развитие деструкции легких во время суперинфекции.
Таким образом, в иммунопатогенезе стафилококковой суперинфекции IL-27, с одной стороны, повышает вероятность развития вторичной бактериальной пневмонии, а с другой — рестриктирует активность воспаления и предупреждает повреждение ткани легкого.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Ahn DS, Parker D, Planet PJ. Secretion of IL-16 through TNFR1 and calpain-caspase signaling contributes to MRSA pneumonia. Mucosal Immunol. 2014 Nov;7(6):1366-74. doi: 10.1038/mi.2014.24.
2. Alijotas-Reig J, Alijotas-Reig J, Esteve-Valverde E, Ferrer-Oliveras R et al. Tumor Necrosis Factor-Alpha and Pregnancy: Focus on Biologics. An Updated and Comprehensive Review. Clin Rev Allergy Immunol. 2017 Jan 4. doi: 10.1007/s12016-016-8596-x.
3. Anderson M, Ohr RJ, Aly KA et al. EssE Promotes Staphylococcus aureus ESS-Dependent Protein Secretion To Modify Host Immune Responses during Infection. J Bacteriol. 2017 Jan 1;199(1). pii: e00527-16. doi: 10.1128%2FJB.00527-16.
4. Archer NK, Adappa ND, Palmer JN et al. Interleukin-17A (IL-17A) and IL-17F Are Critical for Antimicrobial Peptide Production and Clearance of Staphylococcus aureus Nasal Colonization. Infect Immun. 2016 Nov 18;84(12):3575-3583. doi: 10.1128/IAI.00596-16.
5. Archer NK, Harro JM, Shirtliff ME. Clearance of Staphylococcus aureus nasal carriage is T cell dependent and mediated through interleukin-17A expression and neutrophil influx. Infect Immun. 2013 Jun;81(6):2070-5. doi: 10.1128/IAI.00084-13.
6. Aufiero B, Guo M, Young C et al. Staphylococcus aureus induces the expression of tumor necrosis factor-alpha in primary human keratinocytes. Int J Dermatol. 2007 Jul;46(7):687-94. doi: 10.1111/j.1365-4632.2007.03161.x.
7. Ballinger MN, Standiford TJ. Postinfluenza bacterial pneumonia: host defenses gone awry. J Interferon Cytokine Res. 2010 Sep;30(9):643-52. doi: 10.1089/jir.2010.0049.
8. Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009 Nov;10(11):1200-7. doi: 10.1038/ni.1792.
9. Bean AG, Freiberg RA, Andrade S et al. Interleukin 10 protects mice against staphylococcal enterotoxin B-induced lethal shock. Infect Immun. 1993 Nov;61(11):4937-PMID: 8406900. PMC: PMC281261.
10. Blazek K, Eames HL, Weiss M et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med. 2015 Jun 1;212(6):845-53. doi: 10.1084/jem.20140995.
11. Borges da Silva H, Fonseca R, Alvarez JM, D'Império Lima MR. IFN-γ Priming Effects on the Maintenance of Effector Memory CD4(+) T Cells and on Phagocyte Function: Evidences from Infectious Diseases. J Immunol Res. 2015;2015:202816. doi: 10.1155/2015/202816.
12. Bowdridge S, Gause WC. Regulation of alternative macrophage activation by chromatin remodeling. Nat Immunol. 2010 Oct;11(10):879-81. doi: 10.1038/ni1010-879.
Brabcová E, Kolesár L, Thorburn E, Stříž I. Chemokines induced in human respiratory epithelial cells by IL-1 family of cytokines. Folia Biol (Praha). 2014;60(4):180-6. PMID: 25152051.
13. Camporeale A, Poli V. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci (Landmark Ed). 2012 Jun 1;17:2306-26. doi: 10.2741/4054.
14. Capobianchi MR, Uleri E, Caglioti C, Dolei A. Type I IFN family members: similarity, differences and interaction. Cytokine Growth Factor Rev. 2015 Apr;26(2):103-11. doi: 10.1016/j.cytogfr.2014.10.011.
15. Casbon AJ, Long ME, Dunn KW et al. Effects of IFN-γ on intracellular trafficking and activity of macrophage NADPH oxidase flavocytochrome b558. J Leukoc Biol. 2012 Oct;92(4):869-82. doi: 10.1189/jlb.0512244.
16. Chau TA, McCully ML, Brintnell W et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med. 2009 Jun;15(6):641-8. doi: 10.1038/nm.1965.
17. Chen CC, Kobayashi T, Iijima K et al. IL-33 dysregulates regulatory T (Treg) cells and impairs established immunological tolerance in the lungs. J Allergy Clin Immunol. 2017 Feb 11. pii: S0091-6749(17)30228-2. doi: 10.1016/j.jaci.2017.01.015.
18. Chen YG, Zhang Y, Deng LQ et al. Control of Methicillin-Resistant Staphylococcus aureus Pneumonia Utilizing TLR2 Agonist Pam3CSK4. PLoS One. 2016 Mar 14;11(3):e0149233. doi: 10.1371/journal.pone.0149233.
19. Cheng P, Liu T, Zhou WY et al. Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia. BMC Immunol. 2012 Jul 9;13:38. doi: 10.1186/1471-2172-13-38.
20. Choi SM, McAleer JP, Zheng M et al. Innate Stat3-mediated induction of the antimicrobial protein Reg3γ is required for host defense against MRSA pneumonia. J Exp Med. 2013 Mar 11;210(3):551-61. doi: 10.1084/jem.20120260.
21. Chow AW. Adjuvant anti-tumor necrosis factor therapy for staphylococcal arthritis and sepsis: a cautionary note. J Infect Dis. 2011 Aug 1;204(3):332-4. doi: 10.1093/infdis/jir272.
22. Cohen TS, Parker D. Microbial pathogenesis and type III interferons. Cytokine Growth Factor Rev. 2016 Jun;29:45-51. doi: 10.1016/j.cytogfr.2016.02.005.
23. Cohen TS, Prince AS. Bacterial pathogens activate a common inflammatory pathway through IFNλ regulation of PDCD4. PLoS Pathog. 2013;9(10):e1003682. doi: 10.1371/journal.ppat.1003682.
24. Cortez VS, Colonna M. Diversity and function of group 1 innate lymphoid cells. Immunol Lett. 2016 Nov;179:19-24. doi: 10.1016/j.imlet.2016.07.005.
25. Debes GF, Dahl ME, Mahiny AJ et al. Chemotactic responses of IL-4-, IL-10-, and IFN-gamma-producing CD4+ T cells depend on tissue origin and microbial stimulus. J Immunol. 2006 Jan 1;176(1):557-66. doi: 10.4049/jimmunol.176.1.557.
26. Dietert K, Reppe K, Mundhenk L, Witzenrath M, Gruber AD. mCLCA3 modulates IL-17 and CXCL-1 induction and leukocyte recruitment in murine Staphylococcus aureus pneumonia. PLoS One. 2014 Jul 17;9(7):e102606. doi: 10.1371/journal.pone.0102606.
27. Diveu C, McGeachy MJ, Boniface K et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009 May 1;182(9):5748-56. doi: 10.4049/jimmunol.0801162.
28. Doodes PD, Cao Y, Hamel KM et al. IFN-gamma regulates the requirement for IL-17 in proteoglycan-induced arthritis. J Immunol. 2010 Feb 1;184(3):1552-9. doi: 10.4049/jimmunol.0902907.
29. Fox JM, Crabtree JM, Sage LK et al. Interferon Lambda Upregulates IDO1 Expression in Respiratory Epithelial Cells After Influenza Virus Infection. J Interferon Cytokine Res. 2015 Jul;35(7):554-62. doi: 10.1089/jir.2014.0052.
30. Frodermann V, Chau TA, Sayedyahossein S et al. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis. 2011 Jul 15;204(2):253-62. doi: 10.1093/infdis/jir276.
31. Furukawa S, Moriyama M, Miyake K et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. 2017 Feb 13;7:42413. doi: 10.1038/srep42413.
32. Galani IE, Koltsida O, Andreakos E. Type III interferons (IFNs): Emerging Master Regulators of Immunity. Adv Exp Med Biol. 2015;850:1-15. doi: 10.1007/978-3-319-15774-0_1.
33. Gauguet S, D'Ortona S, Ahnger-Pier K et al. Intestinal Microbiota of Mice Influences Resistance to Staphylococcus aureus Pneumonia. Infect Immun. 2015 Oct;83(10):4003-14. doi: 10.1128/IAI.00037-15.
34. Giai C, Gonzalez C, Ledo C et al. Shedding of tumor necrosis factor receptor 1 induced by protein A decreases tumor necrosis factor alpha availability and inflammation during systemic Staphylococcus aureus infection. Infect Immun. 2013 Nov;81(11):4200-7. doi: 10.1128/IAI.00593-13.
35. Gómez MI, Lee A, Reddy B et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004 Aug;10(8):842-8. doi: 10.1038/nm1079.
36. Greenlee-Wacker MC, Nauseef WM. IFN-γ targets macrophage-mediated immune responses toward Staphylococcus aureus. J Leukoc Biol. 2016 Oct 5. pii: jlb.4A1215-565RR. PMC: PMC5295848.
37. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011 Feb;89(2):207-15. doi: 10.1038/icb.2010.158.
38. Hall G, Cullen E, Sawmynaden K et al. Structure of a Potential Therapeutic Antibody Bound to Interleukin-16 (IL-16): mechanistic insights and new therapeutic opportunities. J Biol Chem. 2016 Aug 5;291(32):16840-8. doi: 10.1074/jbc.M115.709303.
39. Held J, Preuße C, Döser A et al. Enhanced acute immune response in IL-12p35-/- mice is followed by accelerated distinct repair mechanisms in Staphylococcus aureus-induced murine brain abscess. J Infect Dis. 2013 Sep 1;208(5):749-60. doi: 10.1093/infdis/jit126.
40. Hernández PP, Mahlakõiv T, Yang I et al. Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. 2015 Jul;16(7):698-707. doi: 10.1038/ni.3180.
41. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008 Dec;226:41-56. doi: 10.1111/j.1600-065X.2008.00707.x.
42. Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009 Oct 16;31(4):539-50. doi: 10.1016/j.immuni.2009.09.002.
43. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015 May;16(5):448-57. doi: 10.1038/ni.3153.
44. Hunter CA, Kastelein R. Interleukin-27: balancing protective and pathological immunity. Immunity. 2012 Dec 14;37(6):960-9. doi: 10.1016/j.immuni.2012.11.003.
45. Huys L, Van Hauwermeiren F, Dejager L et al. Type I interferon drives tumor necrosis factor-induced lethal shock. J Exp Med. 2009 Aug 31;206(9):1873-82. doi: 10.1084/jem.20090213.
46. Ishigame H, Kakuta S, Nagai T et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009 Jan 16;30(1):108-19. doi: 10.1016/j.immuni.2008.11.009.
47. Jacqueline C, Broquet A, Roquilly A et al. Linezolid dampens neutrophil-mediated inflammation in methicillin-resistant Staphylococcus aureus-induced pneumonia and protects the lung of associated damages. J Infect Dis. 2014 Sep 1;210(5):814-23. doi: 10.1093/infdis/jiu145.
48. Joost I, Steinfurt J, Meyer PT et al. Staphylococcus aureus bacteremia with iliac artery endarteritis in a patient receiving ustekinumab. BMC Infect Dis. 2016 Oct 20;16(1):586. doi: 10.1186/s12879-016-1912-5.
49. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016 Jan;12(1):49-62. doi: 10.1038/nrrheum.2015.169.
50. Keck S, Freudenberg M, Huber M. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J Immunol. 2010 May 15;184(10):5809-18. doi: 10.4049/jimmunol.0901423.
51. KleinJan A. Airway inflammation in asthma: key players beyond the Th2 pathway. Curr Opin Pulm Med. 2016 Jan;22(1):46-52. doi: 10.1097/MCP.0000000000000224.
52. Kovarik P, Castiglia V, Ivin M, Ebner F. Type I Interferons in Bacterial Infections: A Balancing Act. Front Immunol. 2016 Dec 26;7:652. doi: 10.3389/fimmu.2016.00652.
53. Kudva A, Scheller EV, Robinson KM et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011 Feb 1;186(3):1666-74. doi: 10.4049/jimmunol.1002194.
54. Labrousse D, Perret M, Hayez D et al. Kineret®/IL-1ra blocks the IL-1/IL-8 inflammatory cascade during recombinant Panton Valentine Leukocidin-triggered pneumonia but not during S. aureus infection. PLoS One. 2014 Jun 6;9(6):e97546. doi: 10.1371/journal.pone.0097546.
55. Lambrecht BN, Hammad H. Asthma: the importance of dysregulated barrier immunity. Eur J Immunol. 2013 Dec;43(12):3125-37. doi: 10.1002/eji.201343730.
56. Lan F, Yuan B, Liu T et al. Interleukin-33 facilitates neutrophil recruitment and bacterial clearance in S. aureus-caused peritonitis. Mol Immunol. 2016 Apr;72:74-80. doi: 10.1016/j.molimm.2016.03.004.
57. Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015 Jul 21;43(1):15-28. doi: 10.1016/j.immuni.2015.07.001.
58. Leech JM, Lacey KA, Mulcahy ME et al. IL-10 Plays Opposing Roles during Staphylococcus aureus Systemic and Localized Infections. J Immunol. 2017 Feb 6. pii: 1601018. doi: 10.4049/jimmunol.1601018.
59. Leemans JC, Vervoordeldonk MJ, Florquin S et al. Differential role of interleukin-6 in lung inflammation induced by lipoteichoic acid and peptidoglycan from Staphylococcus aureus. Am J Respir Crit Care Med. 2002 May 15;165(10):1445-50. doi: 10.1164/rccm.2106045.
60. Li L, Shi QG, Lin F et al. Cytokine IL-6 is required in Citrobacter rodentium infection-induced intestinal Th17 responses and promotes IL-22 expression in inflammatory bowel disease. Mol Med Rep. 2014 Mar;9(3):831-6. doi: 10.3892/mmr.2014.1898.
61. Li Z, Peres AG, Damian AC, Madrenas J. Immunomodulation and Disease Tolerance to Staphylococcus aureus. Pathogens. 2015 Nov 13;4(4):793-815. doi: 10.3390/pathogens4040793.
62. Little FF, Cruikshank WW. Interleukin-16 and peptide derivatives as immunomodulatory therapy in allergic lung disease. Expert Opin Biol Ther. 2004 Jun;4(6):837-46. doi: 10.1517/14712598.4.6.837.
63. Lloyd CM, Saglani S. Epithelial cytokines and pulmonary allergic inflammation. Curr Opin Immunol. 2015 Jun;34:52-8. doi: 10.1016/j.coi.2015.02.001.
64. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001 Feb 23;104(4):487-501. doi: 10.1016/S0092-8674(01)00237-9.
65. Luo Y, Zheng SG. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front Immunol. 2016 Dec 19;7:604. doi: 10.3389/fimmu.2016.00604.
66. Maher BM, Mulcahy ME, Murphy AG et al. Nlrp-3-driven interleukin 17 production by γδT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun. 2013 Dec;81(12):4478-89. doi: 10.1128/IAI.01026-13.
67. Majoros A, Platanitis E, Kernbauer-Hölzl E et al. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol. 2017 Jan 26;8:29. doi: 10.3389/fimmu.2017.00029.
68. Martin FJ, Parker D, Harfenist BS et al. Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect Immun. 2011 May;79(5):1898-904. doi: 10.1128/IAI.01299-10.
69. Martin FJ, Gomez MI, Wetzel DM et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest. 2009 Jul;119(7):1931-9. doi: 10.1172/JCI35879.
70. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015 Feb;15(2):87-103. doi: 10.1038/nri3787.
71. Mendoza Bertelli A, Delpino MV, Lattar S et al. Staphylococcus aureus protein A enhances osteoclastogenesis via TNFR1 and EGFR signaling. Biochim Biophys Acta. 2016 Oct;1862(10):1975-83. doi: 10.1016/j.bbadis.2016.07.016.
72. Mingomataj EÇ, Bakiri AH. Regulator Versus Effector Paradigm: Interleukin-10 as Indicator of the Switching Response. Clin Rev Allergy Immunol. 2016 Feb;50(1):97-113. doi: 10.1007/s12016-015-8514-7.
73. Monticelli LA, Osborne LC, Noti M et al. L-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. 2015 Aug 25;112(34):10762-7. doi: 10.1073/pnas.1509070112.
74. Mordstein M, Michiels T, Staeheli P. What have we learned from the IL28 receptor knockout mouse? J Interferon Cytokine Res. 2010 Aug;30(8):579-84. doi: 10.1089/jir.2010.0061.
75. Mulcahy ME, McLoughlin RM. Staphylococcus aureus and Influenza A Virus: Partners in Coinfection. MBio. 2016 Dec 13;7(6). pii: e02068-16. doi: 10.1128/mBio.02068-16.
76. Mulcahy ME, McLoughlin RM. Host-Bacterial Crosstalk Determines Staphylococcus aureus Nasal Colonization. Trends Microbiol. 2016 Nov;24(11):872-86. doi: 10.1016/j.tim.2016.06.012.
77. Murphy M, Xiong Y, Pattabiraman G, Qiu F, Medvedev AE. Pellino-1 Positively Regulates Toll-like Receptor (TLR) 2 and TLR4 Signaling and Is Suppressed upon Induction of Endotoxin Tolerance. J Biol Chem. 2015 Jul 31;290(31):19218-32. doi: 10.1074/jbc.M115.640128.
78. Nguyen QT, Furuya Y, Roberts S, Metzger DW. Role of Interleukin-12 in Protection against Pulmonary Infection with Methicillin-Resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2015 Oct;59(10):6308-16. doi: 10.1128/AAC.00968-15.
79. Ni G, Wang T, Walton S et al. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. 2015 Feb;293(2):126-9. doi: 10.1016/j.cellimm.2014.12.012.
80. Ning R, Zhang X, Guo X, Li Q. Staphylococcus aureus regulates secretion of interleukin-6 and monocyte chemoattractant protein-1 through activation of nuclear factor kappaB signaling pathway in human osteoblasts. Braz J Infect Dis. 2011 May-Jun;15(3):189-94. PMID: 21670915.
81. Okano M, Fujiwara T, Kariya S et al. Staphylococcal protein A-formulated immune complexes suppress enterotoxin-induced cellular responses in nasal polyps. J Allergy Clin Immunol. 2015 Aug;136(2):343-50.e8. doi: 10.1016/j.jaci.2014.10.058.
82. Ouyang W, Rutz S, Crellin NK et al. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71-109. doi: 10.1146/annurev-immunol-031210-101312.
83. Paczosa MK, Mecsas J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol Mol Biol Rev. 2016 Jun 15;80(3):629-61. doi: 10.1128/MMBR.00078-15.
84. Paolini R, Bernardini G, Molfetta R, Santoni A. NK cells and interferons. Cytokine Growth Factor Rev. 2015 Apr;26(2):113-20. doi: 10.1016/j.cytogfr.2014.11.003.
85. Parker D, Planet PJ, Soong G et al. Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog. 2014 Feb 20;10(2):e1003951. doi: 10.1371/journal.ppat.1003951.
86. Parker D, Ahn D, Cohen T, Prince A. Innate Immune Signaling Activated by MDR Bacteria in the Airway. Physiol Rev. 2016 Jan;96(1):19-53. doi: 10.1152/physrev.00009.2015.
87. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011 Aug;45(2):189-201. doi: 10.1165/rcmb.2011-0011RT.
88. Parker D, Prince A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol. 2012 Oct 15;189(8):4040-6. doi: 10.4049/jimmunol.1201055.
89. Parker D, Prince A. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol. 2011 Dec;32(12):582-8. doi: 10.1016/j.it.2011.09.003.
90. Pesic G, Jeremic J, Nikolic T et al. Interleukin-6 as possible early marker of stress response after femoral fracture. Mol Cell Biochem. 2017 Feb 16. doi: 10.1007/s11010-017-2967-3.
91. Planet PJ, Parker D, Cohen TS et al. Lambda Interferon Restructures the Nasal Microbiome and Increases Susceptibility to Staphylococcus aureus Superinfection. MBio. 2016 Feb 9;7(1):e01939-15. doi: 10.1128/mBio.01939-15.
92. Puimège L, Libert C, Van Hauwermeiren F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. 2014 Jun;25(3):285-300. doi: 10.1016/j.cytogfr.2014.03.004.
93. Reid E, Charleston B. Type I and III interferon production in response to RNA viruses. J Interferon Cytokine Res. 2014 Sep;34(9):649-58. doi: 10.1089/jir.2014.0066.
94. Robinson KM, Choi SM, McHugh KJ et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1β production in mice. J Immunol. 2013 Nov 15;191(10):5153-9. doi: 10.4049/jimmunol.1301237.
95. Robinson KM, Lee B, Scheller EV et al. The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res. 2015 Feb 5;16:10. doi: 10.1186/s12931-015-0168-8.
96. Rose WE, Shukla SK, Berti AD et al. Increased Endovascular Staphylococcus aureus Inoculum is the Link between Elevated Serum IL-10 Concentrations and Mortality in Patients with Bacteremia. Clin Infect Dis. 2017 Feb 16;64(10):1406-12. doi: 10.1093/cid/
cix157.
97. Rubini A. Interleukin-6 and lung inflammation: evidence for a causative role in inducing respiratory system resistance increments. Inflamm Allergy Drug Targets. 2013 Oct;12(5):315-21. doi: 10.2174/1871528111312050003.
98. Rutz S, Ouyang W. Regulation of interleukin-10 and interleukin-22 expression in T helper cells. Curr Opin Immunol. 2011 Oct;23(5):605-12. doi: 10.1016/j.coi.2011.07.018.
99. Rynda-Apple A, Harmsen A, Erickson AS et al. Regulation of IFN-γ by IL-13 dictates susceptibility to secondary postinfluenza MRSA pneumonia. Eur J Immunol. 2014 Nov;44(11):3263-72. doi: 10.1002/eji.201444582.
100. Rynda-Apple A, Dobrinen E, McAlpine M et al. Virus-like particle-induced protection against MRSA pneumonia is dependent on IL-13 and enhancement of phagocyte function. Am J Pathol. 2012 Jul;181(1):196-210. doi: 10.1016/j.ajpath.2012.03.018.
101. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and Bacterial Superinfection: Illuminating the Immunologic Mechanisms of Disease. Infect Immun. 2015 Oct;83(10):3764-70. doi: 10.1128/IAI.00298-15.
102. Sabat R, Grütz G, Warszawska K et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010 Oct;21(5):331-44. doi: 10.1016/j.cytogfr.2010.09.002.
103. Santos-Sierra S, Deshmukh SD, Kalnitski J et al. Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J. 2009 Jul 22;28(14):2018-27. doi: 10.1038/emboj.
2009.158.
104. Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010 Mar;10(3):170-81. doi: 10.1038/nri2711.
105. Satorres SE, Alcaráz LE, Cargnelutti E et al. IFN-gamma plays a detrimental role in murine defense against nasal colonization of Staphylococcus aureus. Immunol Lett. 2009 Apr 27;123(2):185-8. doi: 10.1016/j.imlet.2009.03.003.
106. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011 May;1813(5):878-88. doi: 10.1016/j.bbamcr.2011.01.034.
107. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163-89. doi: 10.1189/jlb.0603252.
108. Shahangian A, Chow EK, Tian X et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009 Jul;119(7):1910-20. doi: 10.1172/JCI35412.
109. Sharma S, Saikia B, Goel S et al. TH17 Cells in STAT3 Related Hyper-IgE Syndrome. Indian J Pediatr. 2016 Oct;83(10):1104-8. doi: 10.1007/s12098-016-2150-y.
110. Shepardson KM, Larson K, Morton RV et al. Differential Type I Interferon Signaling Is a Master Regulator of Susceptibility to Postinfluenza Bacterial Superinfection. MBio. 2016 May 3;7(3). pii: e00506-16. doi: 10.1128/mBio.00506-16.
111. Shukla SK, Rose W, Schrodi SJ. Complex host genetic susceptibility to Staphylococcus aureus infections. Trends Microbiol. 2015 Sep;23(9):529-36. doi: 10.1016/j.tim.2015.05.008.
112. Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004 Mar 15;172(6):3377-81. doi: 10.4049/jimmunol.172.6.3377.
113. Soyka MB, Holzmann D, Basinski TM et al. The Induction of IL-33 in the Sinus Epithelium and Its Influence on T-Helper Cell Responses. PLoS One. 2015 May 1;10(5):e0123163. doi: 10.1371/journal.pone.0123163.
114. Swindle EJ, Brown JM, Rådinger M et al. Interferon-γ enhances both the anti-bacterial and the pro-inflammatory response of human mast cells to Staphylococcus aureus. Immunology. 2015 Nov;146(3):470-85. doi: 10.1111/imm.12524.
115. Tan Y, Kagan JC. Microbe-inducible trafficking pathways that control Toll-like receptor signaling. Traffic. 2017 Jan;18(1):6-17. doi: 10.1111/tra.12454.
116. Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. 2014 Apr;2(4):288-94. doi: 10.1158/2326-6066.CIR-14-0022.
117. Togbe D, Schnyder-Candrian S, Schnyder B et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int J Exp Pathol. 2007 Dec;88(6):387-91. doi: 10.1111/j.1365-2613.2007.00566.x.
118. Tracey KJ, Beutler B, Lowry SF et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986 Oct 24;234(4775):470-4. doi: 10.1126/science.3764421.
119. Tsai HC, Velichko S, Hung LY, Wu R. IL-17A and Th17 cells in lung inflammation: an update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin Dev Immunol. 2013;2013:267971. doi: 10.1155/2013/267971.
120. Ulett GC, Adderson EE. Regulation of Apoptosis by Gram-Positive Bacteria: Mechanistic Diversity and Consequences for Immunity. Curr Immunol Rev. 2006 May;2(2):119-141. doi: 10.2174/157339506776843033.
121. Waters JP, Pober JS, Bradley JR. Tumour necrosis factor in infectious disease. J Pathol. 2013 Jun;230(2):132-47. doi: 10.1002/path.4187.
122. Wu C, Xue Y, Wang P et al. IFN-γ primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR-3473b. J Immunol. 2014 Sep 15;193(6):3036-44. doi: 10.4049/jimmunol.1302379.
123. Ye P, Garvey PB, Zhang P et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001 Sep;25(3):335-40. doi: 10.1165/ajrcmb.25.3.4424.
124. Yin H, Li X, Hu S et al. IL-33 promotes Staphylococcus aureus-infected wound healing in mice. Int Immunopharmacol. 2013 Oct;17(2):432-8. doi: 10.1016/j.intimp.2013.07.008.
125. Yosef N, Shalek AK, Gaublomme JT et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013 Apr 25;496(7446):461-8. doi: 10.1038/nature11981.
126. Zhang X, Shang W, Yuan J et al. Positive Feedback Cycle of TNFα Promotes Staphylococcal Enterotoxin B-Induced THP-1 Cell Apoptosis. Front Cell Infect Microbiol. 2016 Sep 21;6:109. doi: 10.3389/fcimb.2016.00109.
127. Zhang Y, Lv R, Hu X et al. The Role of IL-33 on LPS-Induced Acute Lung Injury in Mice. Inflammation. 2017 Feb;40(1):285-294. doi: 10.1007/s10753-016-0479-z.
128. Zheng Y, Valdez PA, Danilenko DM et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008 Mar;14(3):282-9. doi: 10.1038/nm1720.
129. Zundler S, Neurath MF. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015 Oct;26(5):559-68. doi: 10.1016/j.cytogfr.2015.07.003.
130. Zwirner NW, Ziblat A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front Immunol. 2017 Jan 19;8:25. doi: 10.3389/fimmu.2017.00025.

/130-1.jpg )
/131-1.jpg )
/137-1.jpg )
/139-1.jpg )