Введение
В настоящее время разрабатываются новые антибактериальные средства, нарушающие биосинтез пептидогликана (peptidoglycan — PG), тейхоевых и липотейхоевых кислот, а также прикрепление факторов вирулентности к стенке бактерии. Препараты, нарушающие биосинтез PG, представлены: 1) лекарственными средствами, способными связываться с липидом II; 2) ингибиторами аланин-рацемазы, конверсирующей L-аланин в D-аланин. Медикаментозные средства, препятствующие биосинтезу тейхоевой кислоты (wall teichoicacid — WTA), в основном являются тароцинами, а ингибиторы синтеза липотейхоевой кислоты (lipoteichoic acid — LTA) представлены единственным соединением — компаундом-1771. Средства, нарушающие прикрепление факторов вирулентности к стенке бактерии, представлены ингибиторами сортазы A (sortase A — SrtA) [36, 44, 66].
1. Антибиотики, связывающиеся с липидом II
С учетом того, что липид II является ключевым молекулярным предшественником структурных компонентов клеточной стенки, который находится на поверхности бактериальной клетки, его считают уникальной мишенью для различных антибиотиков (рис. 1), включая гликопептиды (ванкомицин и его аналоги), гликолиподепсипептиды, лантибиотики [36].
Гликопептиды
Среди многочисленных антибиотиков ванкомицин отличается радикально долгим сроком использования в клинической медицине. Так, введенный в клиническую практику еще в 1956 году, он по-прежнему используется для лечения инфекций, вызванных MRSA и полирезистентными стрептококковыми штаммами. Гликопептиды ингибируют синтез стенки грамположительных бактерий, взаимодействуя с терминальной последовательностью d-Ala-d-Ala липида II, что подавляет биосинтез клеточной стенки бактерий. С учетом появления ванкомицинрезистентных бактериальных штаммов разработаны и проектируются аналоги ванкомицина, преодолевающие антибиотикорезистентность грамположительных бактерий (табл. 1) [12].
Антибиотики данной группы, как и оксазолидиноны, отличаются высокой бактерицидной активностью против грамположительных бактерий (табл. 2).
В ряде работ показано, что далбаванцин [15], оритаванцин [14], дипиколил-ванкомицин (dipicolyl-vancomycin) [71], димерная форма ванкомицина [72], телаванцин [11] могут быть использованы при лечении инфекций, вызванных ванкомицинрезистентными грамположительными бактериями.
Далбаванцин
Далбаванцин обладает бактерицидной активностью в отношении грамположительных патогенов, включая Staphylococcus aureus, в том числе MRSA, Streptococcus pneumoniae, Streptococcus agalactiae, Streptococcus pyogenes, Streptococcus anginosus, Enterococcus faecium и Enterococcus faecali [25]. Бактерицидная активность против грамположительных бактерий у далбаванцина в 4–8 раз выше, чем у ванкомицина, и выше, чем у тейкопланина [4, 24]. Среди 1484 мультирезистентных изолятов MRSA общая восприимчивость к далбаванцину составляет 99,8 % [38]. К далбаванцину чувствительны причинно-значимые бактериальные возбудители в 41,3 % случаев при инфекциях нижних дыхательных путей или пневмонии [4], в связи с чем далбаванцин рекомендуется при лечении пневмонии [67].
Дипиколил-ванкомицин
Venkateswarlu Yarlagadda и соавт. [71] для борьбы с ванкомицинрезистентными бактериями (vancomycin-resistant bacteria — VRB) разработали конъюгат дипиколил-ванкомицин (Dipi-van), который характеризуется ингибированием биосинтеза клеточной стенки VRB, активность которой превышает на два порядка уровень подавления синтеза PG ванкомицином.
Оритаванцин
Оритаванцин проявляет высокую бактерицидную активность in vitro против многих грамположительных бактерий, в том числе и против бактерий Staphylococcus aureus, независимо от их восприимчивости к действию метициллина или ванкомицина: МПК50 для hVISA, VISA и VRSA составляют 0,25, 0,5 и 0,5 мг/мл соответственно. Против стрептококков и энтерококков оритаванцин приблизительно в 4–6 раз более активен, чем ванкомицин. Необходимо отметить, что оритаванцин проявляет бактерицидную активность и в присутствии различных генов резистентности к ванкомицину, включая vanA, что отличает его от далбаванцина и телаванцина. Оритаванцин обладает длительным периодом полувыведения — от 200 до 300 часов [34, 41]. Оритаванцин в настоящее время одобрен FDA только для лечения острых бактериальных заболеваний кожи (acute bacterial skin and skin structure infections — ABSSSI). Однако многие исследователи считают, что оритаванцин может быть применен при лечении заболеваний, в том числе и пневмоний, которые вызваны грамположительными патогенами и сопровождаются бактериемией [56, 62, 65]. В то же время, несмотря на наличие достаточного массива данных, свидетельствующих об эффективности оритаванцина in vitro, клинические исследования малочисленны и их результаты не обладают достоверностью, которая позволила бы выработать дифференцированные показания для назначения оритаванцина при VRB-ассоциированных инфекциях [5].
Телаванцин
Телаванцин примерно в 4 раза эффективнее ванкомицина и равноценен по силе бактерицидного действия против стафилококков далбаванцину и оритаванцину [42]. Кроме того, телаванцин проявляет активность против VRB. Средние значения минимальной подавляющей концентрации (МПК) составляют 0,12, 0,25 и 0,5 мг/мл для hVISA, VISA и ванкомицинрезистентных (vancomycin-resistant Staphylococcus aureus — VRSA) клонов Staphylococcus aureus соответственно. Также телаванцин проявляет выраженную бактерицидную активность против Enterococcus spp., включая ванкомицинрезистентные энтерококки (vancomycin-resistant enterococci — VRE) [29]. Согласно данным крупных рандомизированных исследований (III фазы), в которых было проведено сравнение эффективности терапии госпитальной пневмонии телаванцином, вводимым внутривенно в дозе 10 мг/кг каждые 24 часа, и ванкомицином, вводимым внутривенно в дозе 1 грамм каждые 12 часов на протяжении 7–21 суток, вне зависимости от причинно-значимого возбудителя — Staphylococcus aureus, MRSA, ванкомицинрезистентные бактерии — эффективность телаванцина была выше, чем ванкомицина [43]. Считают, что телаванцин (одобренный European Medicines Agency), безусловно, может быть использован при лечении пневмонии, особенно у пациентов с пневмонией, вызванной MRSA. Ali Hassoun и соавт. [19] рекомендуют использовать телаванцин в качестве основного препарата при лечении как внебольничных, так и госпитальных пневмоний, вызванных MRSA. Однако из-за высокого риска нежелательных побочных явлений и потенциальной нефротоксичности препарата требуется осторожность при его клиническом использовании [9].
Гликолиподепсипептиды
Ванкомицин и родственные гликопептиды являются не единственными антибактериальными соединениями, связывающими не ферменты, а субстраты бактериального биосинтеза. Гликолиподепсипептиды, которые являются натуральными продуктами нерибосомальных пептид-синтетаз (nonribosomal peptide synthetase — NRPS), также –могут взаимодействовать с липидом II и ингибировать формирование клеточной стенки бактерии. Натуральные антибактериальные средства, мишенями которых является липид II, представлены лизобактином, эндурацидином A, рамопланином, плюсбацином A3 и тейксобактином (табл. 3). Рамопланин, лизобактин, тейксобактин и другие соединения данной группы связываются с областью липида II, которая включает пирофосфат и первый остаток сахара, но не стеблевой домен пептида [36].
Рамопланин
Рамопланин является первым идентифицированным гликолиподепсипептидным антибиотиком, который получен при ферментации Actinoplanes sp. ATCC 33076 и обладает бактерицидной активностью против MDR-патогенов, включая MRSA и VRE. Однако, демонстрируя высокий уровень антибактериальной активности in vitro, рамопланин характеризуется очень низкой переносимостью при внутривенном введении инфицированным экспериментальным животным. Для преодоления данного ограничения применения рамопланина создаются новые его деривативы [16]. Рамопланин разработан как препарат для перорального применения у пациентов, колонизированных VRE, он также обладает выраженной активностью против Clostridium difficile [40].
В 2017 году Mercedes de la Cruz и соавт. [13] продемонстрировали разнообразие штаммов актиномицетов, которые продуцируют рамопланинподобные соединения, и представили анализ антимикробной активности данных соединений. Авторы считают, что новые аналоги рамопланина, продуцируемые актиномицетами, могут стать основой для разработки новых антибиотиков.
Лизобактин
Лизобактин (катанозин В) продуцируется несколькими родами грамотрицательных глинистых почвенных бактерий. Антибактериальный эффект за счет ингибирования биосинтеза PG впервые был обнаружен в 1987 году. Было установлено, что лизобактин оказывает сильное бактерицидное действие in vitro на MRSA и VRE, а также эффективен при системных стафилококковых и стрептококковых инфекциях у мышей. В 2011 году был идентифицирован кластер генов, участвующих в синтезе лизобактина [18, 21]. Группа авторов под руководством Suzanne Walker [33] в Lysobacter ezymogenes идентифицировала лизобактин, который оказывает выраженное бактерицидное действие против грамположительных бактерий. Гибель клеток, вызванная лизобактином и рамопланином, обусловлена только ингибированием биосинтеза PG. Необходимо отметить, что лизобактин в отличие от рамопланина не оказывает литического действия на человеческие эритроциты. С учетом высокой бактерицидной активности против широкого спектра антибиотикорезистентных патогенов лизобактин может быть перспективным кандидатом для дальнейшего развития антибактериальных препаратов [33].
Плюсбацин A3
Плюсбацин А3 представляет собой циклический липодепсипептид, выделенный из ферментационного бульона Pseudomonas sp. PB-6250, полученной из пробы почвы, собранной в префектуре Окинава (Япония). Молекулярная структура плюсбацина A3 была определена в 1992 году [60]. Плюсбацин А3 ингибирует синтез клеточной стенки грамположительных бактерий, блокируя включение N-ацетилглюкозамина в зарождающийся PG. Согласно результатам исследований Robert D. O’Connor и соавт. [45] плюсбацин А3 обладает двумя механизмами бактерицидного действия. Авторы считают, что плюсбацин А3, во-первых, разрушает ближайший к поверхности мембраны слой PG за счет ингибирования трансгликозилирования и связывания с мономером липида II. Нарушение слоя PG происходит даже в том случае, если отсутствует прямой контакт «хвоста» плюсбацина А3 с липидом II. Возникающая дезорганизация бислоя деполяризует мембрану бактерии, однако без последующего образования пор в клеточной стенке и лизиса бактерий. Во-вторых, вызывает деструкцию бактериальной мембраны за счет высвобождения АТФ из-за нарушения АТФ-связывающих кассетных транспортеров (рис. 2).
/74-1.jpg )
Плюсбацин А3 обладает выраженной антибиотической активностью против MRSA и VRE с МПК от 0,78 до 3,13 мкг/мл. Как и ванкомицин, плюсбацин А3 ингибирует образование зарождающегося пептидогликана, однако, в отличие от ванкомицина, он ингибирует образование промежуточных продуктов липидов, используемых в биосинтезе бактериальных клеточных стенок. Считают, что плюсбацин А3 обладает значительным потенциалом для его применения при лечении инфекций, вызванных VRB [68].
Тейксобактин
Тейксобактин представляет собой новый липидсвязывающий депсипептид, выделенный из β-протобактерии Eleftheria terrae в 2015 году [37]. Тейксобактин характеризуется мощной бактерицидной активностью против многих грамположительных бактерий, включая VRB (МПК тейксобактина колеблется в пределах от 0,25 до 1 мг/мл).
Тейксобактин связывается с липидом II и липидом III, тем самым ингибируя синтез WTA клеточной стенки. Считают, что антибактериальную активность молекулы тейксобактина определяет фрагмент L-аллоэндурацидидина [70]. Данный депсипептид не индуцирует развития резистентности у бактерий. Тейксобактин показал эффективность in vivo при терапии легочной MRSA-ассоциированной инфекции у мышей [66]. Anish Parmar и соавт. [34] создали семь аналогов тейксобактина, которые, возможно, станут молекулярной основой для разработки новых антибиотических средств.
Эндурацидины
Эндурацидины представляют собой редкий структурный класс депсипептидов, представители которого содержат уникальный пятичленный циклический гуанидиновый фрагмент. Впервые эндурацидины как антибиотические средства маннопептимицины были выделены в 2002 году Haiyin He и соавт. [20] из Streptomyces hygroscopicus LL-AC98. Эндурацидины A и B были впервые выделены из Streptomyces fungicidicus B 5477 из образца почвы, взятого в Нисиномии (Япония). Эндурацидины активны против грамположительных бактерий, включая антибиотикорезистентные штаммы, и не проявляют никакой активности против грамотрицательных бактерий (кроме Neisseria gonorrhoeae), грибов или дрожжей. Эндурацидин А и В используют в качестве кормовой добавки антибиотика для свиней и цыплят под торговой маркой Enradin® [1]. Исследования эффективности применения эндурацидинов показали, что они способствуют выздоровлению экспериментальных животных, инфицированных MRSA [55, 63].
2. Ингибиторы аланин-рацемазы
Ингибиторы аланин-рацемазы разрабатываются как противотуберкулезные препараты. Аланин-рацемаза представляет собой фермент, катализирующий конверсию L-аланина в D-аланин, который используется при синтезе PG в организации клеточной стенки бактерий. Поскольку у человека нет известных гомологов, аланин-рацемаза считается важнейшей мишенью для антибактериальных средств. Класс ингибиторов аланин-рацемазы включает в себя O-карбамил-d-серин, D-циклосерин, хлорвинилглицин, алафофин и др. Установлено, что D-циклосерин обладает выраженной активностью против микобактерий, однако его клиническое применение ограниченно из-за выраженной токсичности. Аналоги D-циклосерина и его производные могут стать новым терапевтическим инструментом лечения легочных форм туберкулеза [2].
3. Ингибиторы синтеза тейхоевых кислот
Относительно недавно были идентифицированы ингибиторы ранней стадии синтеза тейхоевых кислот. Данные соединения, получившие название «тароцины», демонстрируют выраженную синергичность бактерицидного действия с широким спектром β-лактамных антибиотиков против различных клинических изолятов MRSA
(табл. 4) [31].
Тароциты и другие ингибиторы синтеза тейхоевых кислот могут стать одним из компонентов рациональной комбинированной терапии с использованием β-лактамных агентов при лечении инфекций, вызванных грамположительными возбудителями, в том числе и MRSA.
4. Ингибиторы синтеза липотейхоевых кислот
Ингибитор 1771 синтазы LTA (LTA synthase — LtaS) [2-оксо-2-(5-фенил-1,3,4-оксадиазол-2-иламино) этил 2-нафто [2,1-b] фуран-1-илацетат] специфически блокирует активность Staphylococcus aureus-LtaS, которая полимеризует 1,3-глицерофосфат в полимеры LTA [58]. Установлено, что компаунд 1771 обладает выраженной бактерицидной активностью не только против бактерий Staphylococcus aureus, но и против MDR энтерококков [52].
5. Ингибиторы сортазы А
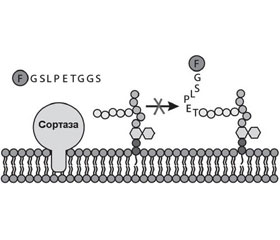
Сортаза А представляет собой мембранный фермент, ответственный за закрепление протеинов на поверхности клеточной стенки грамположительных бактерий. Она катализирует расщепление поверхностного протеина между треониновым и глициновым остатками, что позволяет в последующем карбоксильной группе треонинового остатка поверхностного протеина присоединяться к аминогруппе пентаглицинового мостика PG клеточной мембраны бактерии (рис. 3) [57].
Поверхностные протеины, которые подвергаются обработке SrtA, обладают сигнатурой сигнала сортировки, который содержит LPXTG в C- терминальном регионе молекулы (табл. 5) [7].
Таким образом, SrtA, катализируя закрепление факторов вирулентности на поверхности клеточной стенки стафилококков, энтерококков и стрептококков, играет критическую роль в патогенезе инфекционного процесса, вызванного грамположительными бактериями [6, 10].
Идентифицировано несколько групп химических соединений, подавляющих активность SrtA: 1) натуральные ингибиторы SrtA; 2) синтетические малые молекулы; 3) пептидные дериваты [17].
Натуральные ингибиторы сортазы A
Краткая характеристика натуральных ингибиторов SrtA представлена в табл. 6.
Meng Song и соавт. [61] продемонстрировали, что эпигаллокатехин галлат (EGCG) ингибирует SrtA Streptococcus pneumoniae и способствует выживанию мышей при стрептококковой пневмонии, особенно в ранние сроки после инфицирования. Также авторы показали, что как уровень бактериальной нагрузки в легких, так и степень деструкции легочной ткани инфицированных мышей, леченных EGCG, значительно ниже по сравнению с мышами, не получавшими EGCG.
Синтетические малые молекулы, ингибирующие сортазу A
Класс малых молекул, ингибирующих SrtA, представлен: метил(2E)-2,3-бис(4-метоксифенил)проп-2-еноатом [48]; (Z)-3-(2,5-диметоксифенил)-2-(4-метоксифенил)акрилонитрилом [50]; арил(β-амино)этилкетонами [39]; роданинами [30], пиридазинонами [8, 35] и пиразолтионами [64].
Субстратные дериваты, ингибирующие сортазу A
Ингибиторы SrtA класса пептидных субстратных дериватов представлены аналогами пептидилдиазометана и пептидилхлорметана Cbz (бензилоксикарбонила) — Leu-Pro-Ala-Thr-CHN2 и Cbz-Leu-Pro-Ala-Thr-CH2Cl. Данные ингибиторы ковалентно связывают протеазу, образуя комплекс Михаэлиса, который инактивирует SrtA [59].
По мнению Stella Cascioferro [7], SrtA является идеальной мишенью для препаратов, направленных на подавление активности механизмов вирулентности грамположительных бактерий.
Заключение
Распространение антибиотикорезистентных бактериальных штаммов диктует необходимость разработки новых антибактериальных средств и пересмотра рекомендаций этиологического лечения бактериальных инфекций, в том числе и пневмоний. Для решения проблемы антибиотикорезистентности Европейский союз (инициатива IMI), правительство США (https://www.whitehouse.gov/sites/default/files/docs/national_action_plan_for_combating_antiboticresistant_ bacteria.pdf) и консорциум Wellcome Trust, NIAID (NIH), BARDA и правительство Великобритании — CARB-X (http://www.carb-x.org) выделяют значительные финансовые средства [51], что реализуется во множестве разнообразных научных исследований, в том числе в разработке новых антибиотиков.
Представленные в обзоре новые антибиотики проходят разные фазы исследований. Так, например, тедизолид, далбаванцин уже используются в клинике, а другие антибиотики еще не прошли даже клинических испытаний. По всей вероятности, новые молекулы старых классов антибиотиков и представители новых классов антибиотиков, мишенями которых являются липиды II и III, тейхоевые и липотейхоевые кислоты, аланин-рацемаза и сортаза A, в самом недалеком будущем станут практическими инструментами в клинической практике. Цели и механизмы действия новых антибактериальных соединений предопределяют их клиническую перспективность в будущих стратегиях лечения инфекционных бактериальных заболеваний. Большое количество антибактериальных соединений и разнообразие их механизмов действия дают основание осторожно предположить, что сегодняшние проблемы антибиотикорезистентности, возникающие при лечении внебольничных и нозокомиальных пневмоний, будут в недалеком будущем разрешены.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Atkinson D.J. Enduracididine, a rare amino acid component of peptide antibiotics: Natural products and synthesis / D.J. Atkinson, B.J. Naysmith, D.P. Furkert, M.A. Brimble // Beilstein J. Org. Chem. 2016 Nov 7; 12: 2325-2342. doi: 10.3762 /bjoc.12.226.
2. Azam M.A., Jayaram U. Inhibitors of alanine racemase enzyme: a review // J. Enzyme Inhib. Med. Chem. 2016 Aug; 31(4): 517-26. doi: 10.3109 /14756366.2015.1050010.
3. Bae J. Sesterterpenes from the tropical sponge Coscinoderma sp. / J. Bae, J.E. Jeon, Y.J. Lee et al. // J. Nat. Prod. 2011 Aug 26; 74(8): 1805-11. doi: 10.1021 /np200492k.
4. Biedenbach D.J., Jones R.N. Multicenter evaluation of the in vitro activity of dalbavancin tested against staphylococci and streptococci in 5 European countries: results from the DECIDE Surveillance Program (2007) // Diagn Microbiol Infect Dis. 2009 Jun; 64(2): 177-84. doi: 10.1016 /j.diagmicrobio.2008.12.019.
5. Brade K.D., Rybak J.M., Rybak M.J. Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections // Infect. Dis. Ther. 2016 Mar; 5(1): 1-15. doi: 10.1007 /s40121-016-0103-4.
6. Bradshaw W.J. Molecular features of the sortase enzyme fa–mily / W.J. Bradshaw, A.H. Davies, C.J. Chambers et al. // FEBS J. 2015 Jun; 282(11): 2097-114. doi: 10.1111 /febs.13288.
7. Cascioferro S., Totsika M., Schillaci D. Sortase A: an ideal target for anti-virulence drug development // Microb. Pathog. 2014 Dec; 77: 105-12. doi: 10.1016 /j.micpath.2014.10.007.
8. Chan A.H. NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors / A.H. Chan, S.W. Yi, E.M. Weiner et al. // Chem. Biol. Drug. Des. 2017 Feb 3. doi: 10.1111 /cbdd.12962.
9. Chuan J. Systematic Review and Meta-Analysis of the Efficacy and Safety of Telavancin for Treatment of Infectious Disease: Are We Clearer? / J. Chuan, Y. Zhang, X. He et al. // Front Pharmacol. 2016 Sep 23; 7: 330. doi: 10.3389 /fphar.2016.00330.
10. Clancy K.W., Melvin J.A., McCafferty D.G. Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition // Biopolymers. 2010; 94(4): 385-96. doi: 10.1002 /bip.21472.
11. Corey G.R. Potential role for telavancin in bacteremic infections due to gram-positive pathogens: focus on Staphylococcus aureus / G.R. Corey, E. Rubinstein, M.E. Stryjewski et al. // Clin. Infect. Dis. 2015 Mar 1; 60(5): 787-96. doi: 10.1093 /cid /ciu971.
12. Crotty M.P. New Gram-Positive Agents: the Next Gene–ration of Oxazolidinones and Lipoglycopeptides / M.P. Crotty, T. Krekel, C.A. Burnham, D.J. Ritchie // J. Clin. Microbiol. 2016 Sep; 54(9): 2225-32. doi: 10.1128 /JCM.03395-15.
13. De la Cruz M. Production of Ramoplanin and Ramoplanin Analogs by Actinomycetes / M. de la Cruz, I. González, C.A. Pa–rish et al. // Front. Microbiol. 2017 Mar 6; 8: 343. doi: 10.3389 /fmicb.2017.00343.
14. Duncan L.R. Oritavancin in vitro activity against contemporary Staphylococcus aureus isolates responsible for invasive community- and healthcare-associated infections among patients in the United States (2013-2014) / L.R. Duncan, H.S. Sader, R.K. Flamm et al. // Diagn. Microbiol. Infect. Dis. 2016 Nov; 86(3): 303-306. doi: 10.1016 /j.diagmicrobio.2016.07.025.
15. Esposito S., Bianchini S. Dalbavancin for the treatment of paediatric infectious diseases // Eur. J. Clin. Microbiol. Infect. Dis. 2016 Dec; 35(12): 1895-1901. doi: 10.1007 /s10096-016-2756-z.
16. Fulco P., Wenzel R.P. Ramoplanin: a topical lipoglycodepsipeptide antibacterial agent // Expert Rev. Anti Infect. Ther. 2006 Dec; 4(6): 939-45. doi: 10.1586 /14787210.4.6.939.
17. Gao C. Exploration of multiple Sortase A protein conformations in virtual screening / C. Gao, I. Uzelac, J. Gottfries, L.A. Eriks–son // Sci Rep. 2016 Feb 5; 6: 20413. doi: 10.1038 /srep20413.
18. Hall E.A., Kuru E., VanNieuwenhze M.S. Solid-phase synthesis of lysobactin (katanosin B): insights into structure and function // Org. Lett. 2012 Jun 1; 14(11): 2730-3. doi: 10.1021 /ol300926d.
19. Hassoun A., Friedman B., Saravolatz L.D. Telavancin in the recent Hospital acquired and Ventilator associated pneumonia (HAP /VAP) 2016 guideline // Clin. Infect. Dis. 2017 Mar 24. doi: 10.1093 /cid /cix272.
20. He H. Mannopeptimycins, novel antibacterial glycopeptides from Streptomyces hygroscopicus, LL-AC98 / H. He, R.T. Williamson, B. Shen et al. // J. Am. Chem. Soc. 2002 Aug 21; 124(33): 9729-36.
21. Hou J., Robbel L., Marahiel M.A. Identification and charac–terization of the lysobactin biosynthetic gene cluster reveals mechanistic insights into an unusual termination module architecture // Chem. Biol. 2011 May 27; 18(5): 655-64. doi: 10.1016 /j.chembiol.2011.02.012.
22. Jang K.H. Aaptamines as sortase A inhibitors from the tropical sponge Aaptos aaptos / K.H. Jang, S.C. Chung, J. Shin et al. // Bioorg Med. Chem. Lett. 2007 Oct 1; 17(19): 5366-9. doi: 10.1016 /j.bmcl.2007.08.007.
23. Jeon J.E. Discorhabdins from the Korean marine sponge Sceptrella sp. / J.E. Jeon, Z. Na, M. Jung et al. // J. Nat. Prod. 2010 Feb 26; 73(2): 258-62. doi: 10.1021 /np9005629.
24. Jones R.N. Surrogate analysis of vancomycin to predict susceptible categorization of dalbavancin / R.N. Jones, D.J. Farrell, R.K. Flamm et al. // Diagn. Microbiol. Infect. Dis. 2015 May; 82(1): 73-7. doi: 10.1016 /j.diagmicrobio.2015.01.017.
25. Jones R.N., Schuchert J.E., Mendes R.E. Dalbavancin Activity When Tested against Streptococcus pneumoniae Isolated in Medical Centers on Six Continents (2011 to 2014) // Antimicrob. Agents Chemother. 2016 May 23; 60(6): 3419-25. doi: 10.1128 /AAC.00116-16.
26. Kalil A.C. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: a systematic review and meta-analysis / A.C. Kalil, M.H. Murthy, E.D. Hermsen et al. // Crit. Care Med. 2010 Sep; 38(9): 1802-8. doi: 10.1097 /CCM.0b013e3181eb3b96.
27. Kang S.S. Flavonols inhibit sortases and sortase-mediated Staphylococcus aureus clumping to fibrinogen / S.S. Kang, J.G. Kim, T.H. Lee, K.B. Oh // Biol. Pharm. Bull. 2006 Aug; 29(8): 1751-5. PMID: 16880637.
28. Kim S.H. Inhibition of sortase, a bacterial surface protein anchoring transpeptidase, by beta-sitosterol-3-O-glucopyranoside from Fritillaria verticillata / S.H. Kim, D.S. Shin, M.N. Oh et al. // Biosci Biotechnol. Biochem. 2003 Nov; 67(11): 2477-9. doi: 10.1271 /bbb.67.2477.
29. Klinker K.P., Borgert S.J. Beyond Vancomycin: The Tail of the Lipoglycopeptides // Clin. Ther. 2015 Dec 1; 37(12): 2619-36. doi: 10.1016 /j.clinthera.2015.11.007.
30. Kruger R.G., Dostal P., McCafferty D.G. An economical and preparative orthogonal solid phase synthesis of fluorescein and rhodamine derivatized peptides: FRET substrates for the Staphylococcus aureus sortase SrtA transpeptidase reaction // Chem. Commun. (Camb). 2002 Sep 21; (18): 2092-3. PMID: 12357793.
31. Labroli M.A. Discovery of potent wall teichoic acid early stage inhibitors / M.A. Labroli, J.P. Caldwell, C. Yang et al. // Bioorg. Med. Chem. Lett. 2016 Aug 15; 26(16): 3999-4002. doi: 10.1016 /j.bmcl.2016.06.090.
32. Lee S.H. TarO-specific inhibitors of wall teichoic acid biosynthesis restore β-lactam efficacy against methicillin-resistant staphylococci / S.H. Lee, H. Wang, M. Labroli et al. // Sci Transl. Med. 2016 Mar 9; 8(329): 329ra32. doi: 10.1126 /scitranslmed.aad7364.
33. Lee W. The Mechanism of Action of Lysobactin / W. Lee, K. Schaefer, Y. Qiao et al. // J. Am. Chem. Soc. 2016 Jan 13; 138(1): 100-3. doi: 10.1021 /jacs.5b11807.
34. Lehoux D. Oritavancin Pharmacokinetics and Bone Pe–netration in Rabbits / D. Lehoux, V. Ostiguy, C. Cadieux et al. // Antimicrob. Agents Chemother. 2015 Oct; 59(10): 6501-5. doi: 10.1128 /AAC.00981-15.
35. Li W. Late-stage diversification of biologically active pyri–dazinones via a direct C-H functionalization strategy / W. Li, Z. Fan, K. Geng et al. / Org. Biomol. Chem. 2015 Jan 14; 13(2): 539-48. doi: 10.1039 /c4ob02061h.
36. Ling L.L. A new antibiotic kills pathogens without detectable resistance / L.L. Ling, T. Schneider, A.J. Peoples et al. // Nature. 2015 Jan 22; 517(7535): 455-9. doi: 10.1038 /nature14098.
37. Liu B. Quercitrin, an inhibitor of Sortase A, interferes with the adhesion of Staphylococcal aureus / B. Liu, F. Chen, C. Bi et al. // Molecules. 2015 Apr 13; 20(4): 6533-43. doi: 10.3390 /molecules20046533.
38. Magiorakos A.P. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance / A.P. Magiorakos, A. Srinivasan, R.B. Carey et al. // Clin. Microbiol. Infect. 2012 Mar; 18(3): 268-81. doi: 10.1111 /j.1469-0691.2011.03570.x.
39. Maresso A.W. Activation of inhibitors by sortase triggers irreversible modification of the active site / A.W. Maresso, R. Wu, J.W. Kern et al. // J. Biol. Chem. 2007 Aug 10; 282(32): 23129-39. doi: 10.1074 /jbc.M701857200.
40. Mathur H. The potential for emerging therapeutic options for Clostridium difficile infection / H. Mathur, M.C. Rea, P.D. Cotter et al. // Gut. Microbes. 2014; 5(6): 696-710. doi: 10.4161 /19490976.2014.983768.
41. Mendes R.E. Oritavancin microbiologic features and activity results from the surveillance program in the United States / R.E. Mendes, D.J. Farrell, H.S. Sader, R.N. Jones // Clin. Infect. Dis. 2012 Apr; 54 Suppl 3: S203-13. doi: 10.1093 /cid /cir923.
42. Mendes R.E. Update of the telavancin activity in vitro tested against a worldwide collection of Gram-positive clinical isolates (2013), when applying the revised susceptibility testing method / R.E. Mendes, D.J. Farrell, H.S. Sader et al. // Diagn. Microbiol. Infect. Dis. 2015 Apr; 81(4): 275-9. doi: 10.1016 /j.diagmicrobio.2014.12.011.
43. Nannini E.C., Corey G.R., Stryjewski M.E. Telavancin for the treatment of hospital-acquired pneumonia: findings from the ATTAIN studies // Expert Rev. Anti Infect. Ther. 2012 Aug; 10(8): 847-54. doi: 10.1586 /eri.12.81.
44. Ng V., Chan W.C. New Found Hope for Antibiotic Disco–very: Lipid II Inhibitors // Chemistry. 2016 Aug 26; 22(36): 12606-16. doi: 10.1002 /chem.201601315.
45. O’Connor R.D. Dual Mode of Action for Plusbacin A3 in Staphylococcus aureus / R.D. O’Connor, M. Singh, J. et al. // J. Phys. Chem. B. 2017 Feb Chang 23; 121(7): 1499-1505. doi: 10.1021 /acs.jpcb.6b11039.
46. Oh I. In vitro sortase A inhibitory and antimicrobial activity of flavonoids isolated from the roots of Sophora flavescens / I. Oh, W.Y. Yang, S.C. Chung et al. // Arch. Pharm. Res. 2011 Feb; 34(2): 217-22. doi: 10.1007 /s12272-011-0206-0.
47. Oh K.B. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp / K.B. Oh, W. Mar, S. Kim et al. // Bioorg. Med. Chem. Lett. 2005 Nov 15; 15(22): 4927-31. doi: 10.1016 /j.bmcl.2005.08.021.
48. Oh K.B. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors / K.B. Oh, S.H. Kim, J. Lee et al. // J. Med. Chem. 2004 May 6; 47(10): 2418-21. doi: 10.1021 /jm0498708.
49. Oh K.B. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors / K.B. Oh, M.N. Oh, J.G. Kim et al. // Appl. Microbiol. Biotechnol. 2006 Mar; 70(1): 102-6. PMID: 16010573.
50. Oh K.B. Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model / K.B. Oh, K.W. Nam, H. Ahn et al. // Biochem. Biophys Res. Commun. 2010 May 28; 396(2): 440-4. doi: 10.1016 /j.bbrc.2010.04.113.
51. Outterson K. Accelerating global innovation to address antibacterial resistance: introducing CARB-X / K. Outterson, J.H. Rex, T. Jinks et al. // Nat. Rev. Drug. Discov. 2016 Sep; 15(9): 589-90. doi: 10.1038 /nrd.2016.155.
52. Paganelli F.L. Lipoteichoic acid synthesis inhibition in combination with antibiotics abrogates growth of multidrug-resistant Enterococcus faecium / F.L. Paganelli, T. van de Kamer, E.C. Brouwer et al. // Int. J. Antimicrob. Agents. 2017 Mar; 49(3): 355-363. doi: 10.1016 /j.ijantimicag.2016.12.002.
53. Park B.S. Curcuma longa L. constituents inhibit sortase A and Staphylococcus aureus cell adhesion to fibronectin / B.S. Park, J.G. Kim, M.R. Kim et al. // J. Agric. Food Chem. 2005 Nov 16; 53(23): 9005-9. PMID: 16277395.
54. Parmar A. Defining the molecular structure of teixobactin analogues and understanding their role in antibacterial activities / A. Parmar, S.H. Prior, A. Iyer et al. // Chem Commun (Camb). 2017 Feb 7; 53(12): 2016-2019. doi: 10.1039 /c6cc09490b.
55. Peromet M., Schoutens E., Yourassowsky E. Clinical and microbiological study of enduracidin in infections due to methicillin-resistant strains of Staphylococcus aureus // Chemotherapy. 1973; 19(1): 53-61.PMID: 4490722.
56. Purrello S.M. Methicillin-resistant Staphylococcus aureus infections: A review of the currently available treatment options / S.M. Purrello, J. Garau, E. Giamarellos et al. // J. Glob. Antimicrob. Resist. 2016 Dec; 7: 178-186. doi: 10.1016 /j.jgar.2016.07.010.
57. Rentero Rebollo I. Development of Potent and Selective S. aureus Sortase A Inhibitors Based on Peptide Macrocycles / I. Rentero Rebollo, S. McCallin, D. Bertoldo et al. // ACS Med. Chem. Lett. 2016 Apr 14; 7(6): 606-11. doi: 10.1021 /acsmedchemlett.6b00045.
58. Richter S.G. Small molecule inhibitor of lipoteichoic acid synthesis is an antibiotic for Gram-positive bacteria / S.G. Richter, D. Elli, H.K. Kim et al. // Proc. Natl. Acad. Sci USA. 2013 Feb 26; 110(9): 3531-6. doi: 10.1073 /pnas.1217337110.
59. Scott C.J. Irreversible inhibition of the bacterial cysteine protease-transpeptidase sortase (SrtA) by substrate-derived affinity labels / C.J. Scott, A. McDowell, S.L. Martin et al. // Biochem J. 2002 Sep 15; 366(Pt 3): 953-8. doi: 10.1042 /BJ20020602.
60. Shoji J. Structures of new peptide antibiotics, plusbacins A1-A4 and B1-B4 / J. Shoji, H. Hinoo, T. Katayama et al. // J. Antibiot. (Tokyo). 1992 Jun; 45(6): 824-31. doi: 10.7164 /antibio–tics.45.824.
61. Song M. Epigallocatechin gallate inhibits Streptococcus pneumoniae virulence by simultaneously targeting pneumolysin and sortase A / M. Song, Z. Teng, M. Li et al. // J. Cell. Mol. Med. 2017 Apr 12. doi: 10.1111 /jcmm.13179.
62. Stewart C.L. Real-World Experience with Oritavancin Therapy in Invasive Gram-Positive Infections / Stewart C.L., Turner M.S., Frens J.J. et al. // Infect. Dis. Ther. 2017 Apr 6. doi: 10.1007 /s40121-017-0156-z.
63. Tanayama S., Fugono T., Yamazaki T. Enduracidin, a new antibiotic. IV. The fate of enduracidin administered parente–rally into rabbits // J. Antibiot. (Tokyo). 1968 May; 21(5): 313-9. PMID: 5726287.
64. Uddin R., Lodhi M.U., Ul-Haq Z. Combined pharmacophore and 3D-QSAR study on a series of Staphylococcus aureus Sortase A inhibitors // Chem. Biol. Drug. Des. 2012 Aug; 80(2): 300-14. doi: 10.1111 /j.1747-0285.2012.01403.x.
65. Van Bambeke F. Lipoglycopeptide Antibacterial Agents in Gram-Positive Infections: A Comparative Review // Drugs. 2015 Dec; 75(18): 2073-95. doi: 10.1007 /s40265-015-0505-8.
66. Van Harten R.M. Multidrug-Resistant Enterococcal Infections: New Compounds, Novel Antimicrobial Therapies? / R.M. van Harten, R.J. Willems, N.I. Martin, A.P. Hendrickx // Trends Microbiol. 2017 Feb 13. pii: S0966-842X(17)30016-1. doi: 10.1016 /j.tim.2017.01.004.
67. Welte T., Pletz M.W. Antimicrobial treatment of nosocomial meticillin-resistant Staphylococcus aureus (MRSA) pneumonia: current and future options // Int. J. Antimicrob. Agents. 2010 Nov; 36(5): 391-400. doi: 10.1016 /j.ijantimicag.2010.06.045.
68. Wohlrab A., Lamer R., VanNieuwenhze M.S. Total synthesis of plusbacin A3: a depsipeptide antibiotic active against vancomycin-resistant bacteria // J. Am. Chem. Soc. 2007 Apr 11; 129(14): 4175-7. doi: 10.1021 /ja068455x.
69. Wu M.C. Engineered biosynthesis of enduracidin lipoglycopeptide antibiotics using the ramoplanin mannosyltransferase Ram29 / M.C. Wu, M.Q. Styles, B.J. Law et al. // Microbiology. 2015 Jul; 161(7): 1338-47. doi: 10.1099 /mic.0.000095.
70. Yang H., Chen K.H., Nowick J.S. Elucidation of the Teixo–bactin Pharmacophore // ACS Chem. Biol. 2016 Jul 15; 11(7): 1823-6. doi: 10.1021 /acschembio.6b00295.
71. Yarlagadda V.A. Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria / V. Yarlagadda, P. Sarkar, S. Samaddar, J. Haldar // Angew Chem. Int. Ed. Engl. 2016 Jun 27; 55(27): 7836-40. doi: 10.1002 /anie.201601621.
72. Yarlagadda V. Lipophilic vancomycin aglycon dimer with high activity against vancomycin-resistant bacteria / V. Yarlagadda, P. Sarkar, G.B. Manjunath, J. Haldar // Bioorg. Med. Chem. Lett. 2015 Dec 1; 25(23): 5477-80. doi: 10.1016 /j.bmcl.2015.10.083.

/68-1.jpg )
/69-1.jpg )
/70-1.jpg )
/71-1.jpg )
/72-1.jpg )
/73-1.jpg )
/74-1.jpg )
/75-1.jpg )
/76-1.jpg )
/76-2.jpg)
/77-1.jpg)