Введение
Чрезмерная генерация активированных кислородсодержащих метаболитов (АКМ) и активированных азотсодержащих метаболитов (ААМ) установлена при острых респираторных вирусных инфекциях (ОРВИ). Хронические воспалительные заболевания органов дыхания также сопровождаются усиленным образованием АКМ, ААМ и сниженной функциональной активностью антиоксидантной системы [36].
Острые респираторные вирусные инфекции
Индуцибельная генерация АКМ и ААК — характерный атрибут течения ОРВИ, вызванных вирусами гриппа, парагриппа, респираторно-синцитиальным вирусом, риновирусами и другими вирусами [7, 49]. Вирусы и вирусные РАМР активируют генерацию АКМ и ААК, обусловливая усиление процесса сборки мультипротеинового комплекса НАДФН-оксидазы и экспрессию гена индуцибельной нитрооксидсинтазы (nitric oxide synthase 2 — NOS2) в эпителиальных и провоспалительных клетках респираторного тракта [11]. Внутриклеточные двуцепочечные РНК, образующиеся при репликации вируса, активируют PKR, которая усиливает экспрессию гена NOS2 [43]. Также IFN-γ, секретируемый в слизистой оболочке респираторного тракта в раннем периоде заболевания (от 24 до 36 часов после начала вирусного инфекционного процесса), активирует фактор транскрипции –STAT-1, обусловливая экспрессию NOS2. Результаты экспериментальных исследований подтверждают, что увеличение экспрессии гена NOS2 является одним из основных механизмов противовирусного эффекта процесса активации STAT-1-ассоциированных сигнальных путей эпителиоцитов респираторного тракта человека [49].
Генерируемые АКМ и ААМ оказывают выраженное противовирусное действие [43]. Усиление генерации АКМ и ААК сопровождается постепенным снижением соотношения восстановленного и окисленного глутатиона (GSH/GSSG) и повышением содержания продуктов перекисного окисления липидов, таких как 8-изопростаны, малоновый диальдегид и 4-HNE. Данные изменения свидетельствуют о том, что ОРВИ сопровождаются развитием оксидативного стресса [7, 24]. Также показано, что предшествующий оксидативный стресс, в частности, вызванный выхлопными газами дизельных двигателей, способствует увеличению восприимчивости эпителиоцитов слизистой оболочки респираторного тракта к инфицированию вирусами гриппа и сопровождается усилением репликации вируса гриппа. Однако выраженность репликации вирусов на фоне оксидативного стресса прямо коррелирует с уровнем продукции IFN-β, IFN-β-ассоциированной экспрессии протеина MXA и обратно пропорционально связана со степенью продукции IFN-γ макрофагами. Ilona Jaspers и соавт. [20] предполагают, что АКМ, не подавляя активность продукции IFN-β, редуцируют способность IFN-β-ассоциированного ответа вызывать ингибирование репликации вируса гриппа. Поскольку введение GSH способствует снижению активности вирусной репликации, считают, что нарушение функционирования глутатионовой системы может быть одной из причин тяжелого течения ОРВИ. Высокий уровень GSH ингибирует репликацию как вируса гриппа, так и вируса простого герпеса [45]. Известно, что риновирусная инфекция сопровождается усилением экспрессии ICAM-1, которая является рецептором для 90 % различных серологических типов риновирусов. Показано, что риновирусная инфекция приводит к снижению уровня внутриклеточной концентрации восстановленного глутатиона, в то время как повышение уровня восстановленного глутатиона сопровождается ингибированием экспрессии ICAM-1 и снижением активности риновирусной инфекции [33].
Усиление активности механизмов антиоксидантной защиты происходит в раннем периоде ОРВИ — уже через 6 часов после заражения вирусом наблюдается увеличение экспрессии Mn-супероксиддисмутазы (Mn superoxide dismutase — MnSOD), каталазы (catalase — CAT), глутатионпероксидазы (glutathione peroxidase — GPX) и глутатион-S-трансферазы (glutathione S-transferase — GST) [17, 18]. Глобальное исследование экспрессии генов в эпителиоцитах дыхательных путей человека показало, что за активацией механизмов антиоксидантной защиты раннего периода ОРВИ следует быстрое снижение активности большинства ферментов антиоксидантной системы: каталазы, GPX, GST и пероксиредоксинов (peroxiredoxin — PRDX) [7]. Установлено, что респираторно-тропные вирусы способны ингибировать активность фактора транскрипции NRF2 (nuclear factor, erythroid 2 like 2), тем самым снижая экспрессию антиоксидантных ферментов [12, 19]. Прогрессивное снижение уровня содержания каталазы и GST наблюдается и в жидкости бронхоальвеолярного лаважа. Исключение составляют
Cu/ZnSOD и MnSOD. Уровень активности
Cu/ZnSOD практически не меняется в различные периоды болезни, а экспрессия MnSOD характеризуется постепенным увеличением на всем протяжении заболевания [1, 42]. По всей вероятности, вирусы гриппа, парагриппа и респираторно-синцитиальные вирусы индуцируют транскрипционную активность гена MnSOD [19]. Увеличение каталитической активности MnSOD в сочетании с прогрессирующим снижением экспрессии каталазы, GPX, GST и PRDX создает определенную ситуацию, которая характеризуется достаточным уровнем димутации супер–оксид анион-радикала с образованием перекиси водорода (H2O2) при недостаточности активности ее инактивации. Вероятно, при ОРВИ накопление именно внутриклеточной H2O2 предопределяет окислительно-восстановительный статус и вероятность повреждения структурных элементов клетки (рис. 1).
/120-1.jpg )
Установлено, что повышение внутриклеточной активности SOD в ответ на инфицирование респираторно-синцитиальным вирусом влияет на модуляцию АКМ-ассоциированных сигнальных путей и последующую вирусно-индуцированную экспрессию генов, в то время как повышение уровня каталазы и/или активности пероксидазы ингибирует экспрессию провоспалительных генов. Таким образом, медикаментозное усиление активности каталазы и пероксидаз может быть эффективным методом предупреждения развития оксидативного стресса при ОРВИ [18]. Применение доноров NO или индукторов iNOS ингибирует репликацию вирусов и является перспективным направлением для разработки новых противовирусных методов лечения [49].
Хронические воспалительные заболевания органов дыхания
В многочисленных экспериментальных и клинических научных работах показано, что снижение уровня содержания GSH в бронхоальвеолярной жидкости сопровождает острый респираторный дистресс-синдром, коррелирует с тяжестью течения муковисцидоза. Считают, что низкий уровень GSH у пациентов с воспалительными заболеваниями органов дыхания способствует пролонгации процесса воспаления и развитию фиброзирования. Однако в бронхоальвеолярной жидкости у пациентов с легкой тяжестью бронхиальной астмы наблюдалось увеличение общего содержания глутатиона [9, 25].
Показано, что уровень внутриклеточного содержания GSH предопределяет пролиферацию Т-лимфоцитов, дифференциацию наивных CD4+-Т-клеток. Снижение уровня содержания GSH в антигенпрезентирующих клетках подав–ляет продукцию Th1-ассоциированных цитокинов (IFN-γ, –IL-12) и способствует развитию Th2-ассоциированного гуморального иммунного ответа, который сопровождается синтезом IgG1, в то время как антигенпрезентирующие клетки с высоким внутриклеточным уровнем содержания GSH секретируют цитокины, способствующие дифференциации Th1-клеток и продукции IgG2a и IgG2b. Влияние IFN-g обусловливает увеличение уровня внутриклеточного содержания GSH, а действие IL-4 приводит к снижению внутриклеточной концентрации GSH. Введение глутатиона мышам приводит к увеличению продукции IFN-g, IL-27 и ингибированию синтеза IL-13 T-клетками. –Таким образом, GSH предопределяет развитие Th1-ассоциированного иммунного ответа, а его дефицит — Th2-ассоциированного иммунного ответа. Лекарственные средства, снижающие уровень содержания глутатиона (этанол, циклофосфамид, ацетаминофен), ассоциированы с развитием Th2-зависимых заболеваний, в том числе и бронхиальной астмы [4, 6, 14, 21]. Hee Yeon Won и соавт. [48] продемонстрировали, что GPX1-дефицитные Т-лимфоциты отличаются более высоким уровнем внутриклеточного содержания глутатиона, продукции АКМ, секреции IL-2 и выраженной склонностью к дифференциации в Th1- и Th17-клетки. Мыши с нокаутом гена GPx1 были защищены от аллергической реакции на овальбумин, которая наблюдалась у диких мышей в виде эозинофильной инфильтрации, гиперплазии бокаловидных клеток и гиперчувствительности бронхиального дерева.
У пациентов с бронхиальной астмой и хронической обструктивной болезнью легких (ХОБЛ) наблюдается снижение активности SOD, каталазы и GST как в жидкости бронхоальвеолярного лаважа, так и в эпителиоцитах дыхательных путей [15, 29, 41]. АКМ и ААМ принимают участие в хронизации процесса, поддерживая активность и формируя нозоспецифичность процесса воспаления, а также способствуя развитию фиброза легочной ткани.
Оксидативный стресс и воспаление
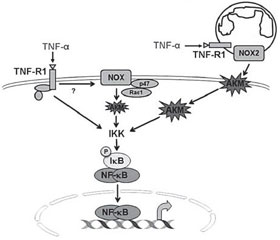
Оксидативный стресс и воспаление являются сопряженными процессами, особенно в респираторном тракте, ткань которого является высоко-АКМ-зависимой. В частности, возбуждение NF-κB, который является одним из ключевых провоспалительных факторов транскрипции, может быть индуцировано NOX-АКМ-зависимым и NOX-АКМ-независимым способом. Однако в легочной ткани NOX-АКМ-зависимые пути активации –NF-κB играют основную роль в индукции синтеза провоспалительных цитокинов (рис. 2) [31, 50].
АКМ, оказывая влияние на рецепторы, компоненты внутриклеточных сигнальных путей и факторы транскрипции, индуцируют продукцию провоспалительных цитокинов. В зависимости от заболевания одни и те же цитокины могут выполнять как пато-, так и протекторную роль. Для бронхиальной астмы и ХОБЛ характерны нозоспецифические особенности развития воспалительного процесса бронхиального дерева. При бронхиальной астме наблюдается преимущественно Th2-ответ, а при ХОБЛ — Th1-ответ.
Хронический воспалительный процесс респираторного тракта сопровождается увеличением не только генерации ААМ, но и продукции асимметричного NG,NG-диметил-L-аргинина (ADMA), который является естественным ингибитором NOS. Эпителиоциты слизистой оболочки респираторного тракта являются основными продуцентами ADMA. Показано, что при воспалении респираторного тракта повышается активность протеин-аргинина метилтрансферазы I типа PRMT2, которая, метилируя аргининовые остатки, определяет образование ADMA, и снижается активность NG,NG-диметиларгинина диметиламиногидролазы (DDAH2), которая метаболизирует ADMA в цитруллин и диметиламин. Асимметричный NG,NG-диметил-L-аргинин конкурентно ингибирует NOS, вытесняя L-аргинин. Повышение уровня концентрации ADMA ведет к снижению генерации NO и увеличению продукции O2–• и пероксинитрита. Однако ADMA существенно ингибирует генерацию NO только nNOS и eNOS и очень слабо подавляет функциональную активность iNOS, также ADMA индуцирует высвобождение эндотелиоцитами ангиотензина II, который, в свою очередь, активирует генерацию O2–• НАДФH-оксидазой. В связи с этим высокая концентрация ADMA может усилить и пролонгировать проявления нитрозативного стресса. В состоянии практического здоровья концентрация ADMA в сыворотке крови людей колеблется на уровне 0,35–0,70 мкмоль/л [23, 35, 44]. При бронхиальной астме и ХОБЛ наблюдается высокий уровень продукции ADMA [3, 32]. Избыток ADMA индуцирует синтез коллагена в респираторном тракте, обусловливая ремоделирование бронхиальных стенок, способствует развитию эндотелиальной дисфункции и несет риск развития сердечно-сосудистых заболеваний [2, 30].
АМК-ассоциированный путь развития фиброза легочной ткани
На протяжении многих лет накапливались доказательства того, что оксидативный стресс играет важную роль в патогенезе легочного фиброза. Установлено, что АКМ могут активировать некоторые гены, которые связаны с ростом, гибелью клеток и пролиферацией фибробластов. Установлено, что GSH ингибирует, а GSSG не влияет на пролиферацию фибробластов легочной ткани. Поэтому состояния, которые сопровождаются снижением уровня GSH, сопровождаются гиперпролиферацией фибробластов. Сниженный уровень GSH приводит к увеличению продукции TGF-β1 и коллагена [27, 40]. TGF-β1 — это цитокин, который обладает выраженными противовоспалительным и профибротическим свойствами. В ткани легкого основными продуцентами TGF-β1 являются эпителиоциты, фибробласты, альвеолярные и тканевые макрофаги. Индукторами синтеза TGF-β1 считают: диоксид кремния, блеомицин, гипероксии и паракват. Профибротическое действие TGF-β1 преимущественно реализуется через внутриклеточный сигнальный путь, ассоциированный с транскрипционным фактором Smad3 (рис. 3).
/122-1.jpg )
TGF-β1 является основным фактором, который несет ответственность за дифференцировку фибробластов. Данный цитокин реализует свое действие через внутриклеточные Smad-ассоциированные сигнальные пути с участием митогенактивируемой протеинкиназы и протеинкиназы B. Активация TGF-β1/Smad3-пути в фибробластах обусловливает их трансдифференцировку в миофибробласты и индукцию экспрессии: фибронектина, коллагенов, фактора роста соединительной ткани, тромбоцитарного фактора роста, ингибитора активатора плазминогена 1 и др. Миофибробласты являются специализированными клетками, определяющими развитие фиброза ткани. Возбуждение TGF-β1/Smad3-пути в эпителиальных клетках приводит к индукции синтеза коллагена и активации эпителиально-мезенхимального перехода — явления, которое в настоящее время считается ключевым в развитии фиброза легких. Учитывая, что активация PPAR-g ингибирует проявления эффектов TGF-β1/Smad3-сигнального пути, считают, что агонисты PPAR-g могут быть использованы при лечении процессов, сопровождающихся развитием фиброза легкого [8, 22, 26, 37, 39].
Одним из ранних эффектов действия TGF-β1 является усиление генерации H2O2 фибробластами легочной ткани. Meghna Waghray и соавт. [46] впервые установили наличие генерации H2O2 мощностей фибробластами, которые были получены от взрослых пациентов с фиброзирующим альвеолитом. Авторами было показано, что уровень генерации H2O2 настолько высок, что может вызвать гибель эпителиальных клеток слизистой оболочки мелких бронхов. H2O2-генерирующие фибробласты характеризуются специфическим, устойчивым к агрессивному действию АКМ фенотипом. В последнее время установлено, что TGF-β1 селективно индуцирует экспрессию изоформы НАДФH-оксидазы NOX4 в мезенхимальных клетках, в том числе гладкомышечных клетках легочной артерии, фибробластах сердечной, почечной, легочной тканей. –TGF-β1-индуцированная экспрессия NOX4 тесно связана с дифференцировкой фибробластов. Повышенная генерация супероксид-аниона радикала NOX4 сопровождается индукцией синтеза α-актина гладкомышечных клеток и протеинов экстрацеллюлярного матрикса (фактор роста соединительной ткани, фибронектин, коллаген) и дифференцировкой фибробластов в миофибробласты (рис. 4) [5, 10, 13, 16].
/122-2.jpg )
Charalambos Michaeloudes и соавт. [28] показали, что под влиянием TGF-β1 активируется Smad-сигнальный путь, который ведет к индукции экспрессии NOX4 и IL-6 и ингибиции экспрессии каталазы и MnSOD. В результате действия АКМ возбуждается и PI3K, которая также способствует экспрессии NOX4 и IL-6. Усиление активности NOX4 может индуцировать экспрессию MnSOD, но не каталазы или IL-6.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Afolayan A.J. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn / A.J. Afolayan, A. Eis, R.J. Teng et al. // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2012, Nov 15. — 303(10). — Р. 870-9. — doi: 10.1152/ajplung.00098.2012.
2. Ahmad T. Altered asymmetric dimethyl arginine metabolism in allergically inflamed mouse lungs / T. Ahmad, U. Mabalirajan, B. Ghosh, A. Agrawal // Am. J. Respir. Cell. Mol. Biol. — 2010 Jan. — 42(1). — Р. 3-8. — doi: 10.1165/rcmb.2009-0137RC.
3. Ahmad T. Simvastatin improves epithelial dysfunction and airway hyperresponsiveness: fromasymmetric dimethyl-arginine to asthma / Ahmad T., Mabalirajan U., Sharma A. et al. // Am. J. Respir. Cell. Mol. Biol. — 2011 Apr. — 44(4). — Р. 531-9. — doi: 10.1165/rcmb.2010-0041OC.
4. Allmers H., Skudlik C., John S.M. Acetaminophen use: a risk for asthma? // Curr. Allergy Asthma Rep. — 2009 Mar. — 9(2). — Р. 164-7. — PMID: 19210907.
5. Amara N. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts / N. Amara, D. Goven, F. Prost et al. // Thorax. — 2010 Aug. — 65(8). — Р. 733-8. — doi: 10.1136/thx.2009.113456.
6. Ballatori N. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology / N. Ballatori, S.M. Krance, R. Marchan, C.L. Hammond // Mol. Aspects Med. — 2009 Feb-Apr. — 30(1–2). — Р. 13-28. — doi: 10.1016/j.mam.2008.08.004.
7. Bao X. Identification of human metapneumovirus-induced gene networks in airway epithelial cells by microarray analysis / X. Bao, M. Sinha, T. Liu et al. // Virology. — 2008, Apr 25. — 374(1). — Р. 114-27. — doi: 10.1016/j.virol.2007.12.024.
8. Biernacka A., Dobaczewski M., Frangogiannis N.G. TGF-β signaling in fibrosis // Growth Factors. — 2011 Oct. — 29(5). — Р. 196-202. — doi: 10.3109/08977194.2011.595714.
9. Biswas S.K., Rahman I. Environmental toxicity, redox signa–ling and lung inflammation: the role of glutathione // Mol. Aspects Med. — 2009 Feb-Apr. — 30(1–2). — Р. 60-76. — doi: 10.1016/j.mam.2008.07.001.
10. Carnesecchi S. A key role for NOX4 in epithelial cell death during development of lung fibrosis / S. Carnesecchi, C. Deffert, Y. Donati et al. // Antioxid Redox Signal. — 2011, Aug 1. — 15(3). — Р. 607-19. — doi: 10.1089/ars.2010.3829.
11. Castro S.M. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation / S.M. Castro, A. Guerrero-Plata, G. Suarez-Real et al. // Am. J. Respir. Crit. Care Med. — 2006, Dec 15. — 174(12). — Р. 1361-9. — doi: 10.1164/rccm.200603-319OC.
12. Cho H.Y., Kleeberger S.R. Association of Nrf2 with airway pathogenesis: lessons learned from genetic mouse models // Arch. Toxi–col. — 2015 Nov. — 89(11). — Р. 1931-57. — doi: 10.1007/s00204-015-1557-y.
13. Crestani B., Besnard V., Boczkowski J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis // Int. J. Biochem. Cell. Biol. — 2011 Aug. — 43(8). — Р. 1086-9. — doi: 10.1016/j.biocel.2011.04.003.
14. Fraternale A. The increase in intra-macrophage thiols induced by new pro-GSH molecules directs the Th1 skewing in ovalbumin immunized mice / A. Fraternale, M.F. Paoletti, S. Dominici et al. // Vaccine. — 2010, Nov 10. — 28(48). — Р. 7676-82. — doi: 10.1016/j.vaccine.2010.09.033.
15. Ghosh S. Nitrotyrosine proteome survey in asthma identifies oxidative mechanis of catalase inactivation / S. Ghosh, A.J. Janocha, M.A. Aro–nica et al. // J. Immunol. — 2006, May 1. — 176(9). — Р. 5587-97.
16. Hecker L., Cheng J., Thannickal V.J. Targeting NOX enzymes in pulmonary fibrosis // Cell. Mol. Life Sci. — 2012 Jul. — 69(14). — Р. 2365-71. — doi: 10.1007/s00018-012-1012-7.
17. Hosakote Y.M. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus / Y.M. Hosakote, N. Komaravelli, N. Mautemps et al. // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2012, Dec 1. — 303(11). — Р. 991-1000. — doi: 10.1152/ajplung.00192.2012.
18. Hosakote Y.M. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes / Y.M. Hosakote, T. Liu, S.M. Castro et al. // Am. J. Respir. Cell. Mol. Biol. — 2009 Sep. — 41(3). — Р. 348-57. — doi: 10.1165/rcmb.2008-0330OC.
19. Hosakote Y.M. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis / Y.M. Hosakote, P.D. Jantzi, D.L. Esham et al. // Am. J. Respir. Crit. Care Med. — 2011, Jun 1. — 183(11). — Р. 1550-60. — doi: 10.1164/rccm.201010-1755OC.
20. Jaspers I. Diesel exhaust enhances influenza virus infections in respiratory epithelial cells / I. Jaspers, J.M. Ciencewicki, W. Zhang et al. // Part Fibre Toxicol. 2009, Aug 14. — 6. — Р. 22. — doi: 10.1186/1743-8977-6-22.
21. Kamide Y. Intracellular glutathione redox status in human dendritic cells regulates IL-27 production and T-cell polarization / Kamide Y., Utsugi M., Dobashi K. et al. // Allergy. — 2011 Sep. — 66(9). — Р. 1183-92. — doi: 10.1111/j.1398-9995.2011.02611.x.
22. Kis K., Liu X., Hagood J.S. Myofibroblast differentiation and survival in fibrotic disease // Expert Rev. Mol. Med. — 2011, Aug 23. — 13. — Р. 27. — doi: 10.1017/S1462399411001967.
23. Klein E. Asymmetric dimethylarginine potentiates lung inflammation in a mouse model of allergic asthma / Klein E., Weigel J., Buford M.C. et al. // Am. J. Physiol. Lung Cell. Mol. Physiol. — 2010 Dec. — 299(6). — Р. 816-25. — doi: 10.1152/ajplung.00188.201.
24. Komaravelli N. Respiratory syncytial virus infection down-re–gulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2 / N. Komaravelli, B. Tian, T. Ivan–ciuc et al. // Free Radic. Biol. Med. — 2015 Nov. — 88(Pt B). — Р. 391-403. — doi: 10.1016/j.freeradbiomed.2015.05.043.
25. Kurundkar A., Thannickal V.J. Redox mechanisms in age-related lung fibrosis // Redox Biol. — 2016 Oct. — 9. — Р. 67-76. — doi: 10.1016/j.redox.2016.06.005.
26. Lakshmi S.P., Reddy A.T., Reddy R.C. Transforming growth factor β suppresses peroxisome proliferator-activated receptor γ expression via both SMAD binding and novel TGF-β inhibitory elements // Biochem. J. — 2017, Apr 24. — 474(9). — Р. 1531-1546. — doi: 10.1042/BCJ20160943.
27. Liu R.M. Transforming growth factor β suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model / Liu R.M., Vayalil P.K., Ballinger C. et al. // Free Radic. Biol. Med. — 2012, Aug 1. — 53(3). — Р. 554-63. — doi: 10.1016/j.freeradbiomed.2012.05.016.
28. Michaeloudes C. TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells / C. Michaeloudes, M.B. Sukkar, N.M. Khorasani et al. // Am. J. Physiol. Lung Cell. Mol. Physiol. — 2011 Feb. — 300(2). — Р. 295-304. — doi: 10.1152/ajplung.00134.2010.
29. Misso N.L., Thompson P.J. Oxidative stress and antioxidant deficiencies in asthma: potential modification by diet // Redox Rep. — 2005. — 10(5). — Р. 247-55. — doi: 10.1179/135100005X70233.
30. Mohan S., Fung H.L. Mechanism of cellular oxidation stress induced by asymmetric dimethylarginine // Int. J. Mol. Sci. — 2012. — 13(6). — Р. 7521-31. — doi: 10.3390/ijms13067521.
31. Mroz R.M. Molecular basis of chronic inflammation in lung diseases: new therapeutic approach / R.M. Mroz, J. Noparlik, E. Chyczewska et al. // J. Physiol. Pharmacol. — 2007 Nov. — 58, Suppl. 5(Pt 2). — Р. 453-60. — PMID: 18204158.
32. Nural S. Inflammatory processes and effects of continuous po–sitive airway pressure (CPAP) in overlap syndrome / Nural S., Günay E., Halici B. et al. // Inflammation. — 2013 Feb. — 36(1). — Р. 66-74. — doi: 10.1007/s10753-012-9520-z.
33. Papi A. Reducing agents inhibit rhinovirus-induced up-regu–lation of the rhinovirus receptor intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells / A. Papi, N.G. Papadopoulos, L.A. Stanciu et al. // FASEB J. — 2002 Dec. — 16(14). — Р. 1934-6. — doi: 10.1096/fj.02-0118fje.
34. Perrone L.A. Inducible nitric oxide contributes to viral pathogenesis following highly pathogenic influenza virus infection in mice / L.A. Perrone, J.A. Belser, D.A. Wadford et al. // J. Infect. Dis. — 2013, May 15. — 207(10). — Р. 1576-84. — doi: 10.1093/infdis/jit062.
35. Pope A.J., Karuppiah K., Cardounel A.J. Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production // Pharmacol. Res. — 2009 Dec. — 60(6). — Р. 461-5. — doi: 10.1016/j.phrs.2009.07.016.
36. Rahman I. Oxidant and antioxidant balance in the airways and airway diseases / I. Rahman, S.K. Biswas, A. Kode // Eur. J. Pharmacol. — 2006, Mar 8. — 533(1–3). — Р. 222-39. — doi: 10.1016/j.ejphar.2005.12.087.
37. Teoh C.M., Tan S.S., Tran T. Integrins as Therapeutic Targets for Respiratory Diseases // Curr. Mol. Med. — 2015. — 15(8). — Р. 714-34. — PMID: 26391549.
38. Thannickal V.J. Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase // Fibrogenesis Tissue Repair. — 2012, Jun 6. — 5(Suppl. 1). — Р. 23. — doi: 10.1186/1755-1536-5-S1-S23.
39. Thomas B.J. In the Shadow of Fibrosis: Innate Immune Suppression Mediated by Transforming Growth Factor-β / B.J. Thomas, O.K. Kan, K.L. Loveland et al. // Am. J. Respir. Cell. Mol. Biol. — 2016 Dec. — 55(6). — Р. 759-766. — PMID: 27603223.
40. Todd N.W., Luzina I.G., Atamas S.P. Molecular and cellular mechanisms of pulmonary fibrosis // Fibrogenesis Tissue Repair. — 2012, Jul 23. — 5(1). — Р. 11. — doi: 10.1186/1755-1536-5-11.
41. Tomaki M. Decreased expression of antioxidant enzymes and increased expression of chemokines in COPD lung / M. Tomaki, H. Sugiura, A. Koarai et al. // Pulm. Pharmacol. Ther. — 2007. — 20(5). — Р. 596-605. — Epub. 2006, Jul 11. — doi: 10.1016/j.pupt.2006.06.006.
42. Umasuthan N. Characterization of rock bream (Oplegnathus fasciatus) cytosolic Cu/Zn superoxide dismutase in terms of molecular structure, genomic arrangement, stress-induced mRNA expression and antioxidant function / N. Umasuthan, S.D. Bathige, W.S. Thula–sitha et al. // Comp. Biochem. Physiol. B Biochem. Mol. Biol. — 2014 Oct. — 176. — Р. 18-33. — doi: 10.1016/j.cbpb.2014.07.004.
43. Vareille M. The airway epithelium: soldier in the fight against respiratory viruses / M. Vareille, E. Kieningr, M.R. Edwards, N. Regamey // Clin. Microbiol. Rev. — 2011 Jan. — 24(1). — Р. 210-29. — doi: 10.1128/CMR.00014-10.
44. Veresh Z. ADMA impairs nitric oxide-mediated arteriolar function due to increased superoxide production by angiotensin –II-NAD(P)H oxidase pathway / Z. Veresh, A. Racz, G. Lotz, A. Koller // Hypertension. — 2008 Nov. — 52(5). — Р. 960-6. — doi: 10.1161/HYPERTENSIONAHA.108.116731.
45. Vogel J.U. Effects of S-acetylglutathione in cell and animal model of herpes simplex virus type 1 infection / J.U. Vogel, J. Cinatl, N. Dauletbaev et al. // Med. Microbiol. Immunol. — 2005 Jan. — 194(1–2). — Р. 55-9. — doi: 10.1007/s00430-003-0212-z.
46. Waghray M. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts / M. Waghray, Z. Cui, J.C. Horowitz et al. // FASEB J. — 2005 May. — 19(7). — Р. 854-6. — Epub. 2005, Feb 16. — doi: 10.1096/fj.04-2882fje.
47. Weiss C.H. Poteasomal regulation of pulmonary fibrosis / C.H. Weiss, G.R. Budinger, G.M. Mutlu, M. Jain // Proc. Am. Thorac. Soc. — 2010 Feb. — 7(1). — Р. 77-83. — doi: 10.1513/pats.200906-055JS.
48. Won H.Y. Glutathione peroxidase 1 deficiency attenuates allergen-induced airway inflammation by suppressing Th2 and Th17 cell development / Won H.Y., Sohn J.H., Min H.J. et al. // Antioxid. Redox Signal. — 2010, Sep 1. — 13(5). — Р. 575-87. — doi: 10.1089/ars.2009.2989.
49. Xu W. Role of epithelial nitric oxide in airway viral infection / W. Xu, S. Zheng, R.A. Dweik, S.C. Erzurum // Free Radic. Biol. Med. — 2006, Jul 1. — 41(1). — Р. 19-28. — doi: 10.1016/j.freeradbiomed.2006.01.037.
50. Zhang W.J. Genetic ablation of phagocytic NADPH oxidase in mice limits TNFα-induced inflammation in the lungs but not other tissues / W.J. Zhang, H. Wei, Y.T. Tien, B. Frei // Free Radic. Biol. Med. — 2011, Jun 1. — 50(11). — Р. 1517-25. — doi: 10.1016/j.freeradbiomed.2011.02.027.

/120-1.jpg )
/121-1.jpg )
/122-1.jpg )
/122-2.jpg )