Проблема діагностики та лікування орфанних захворювань є важливим викликом для сучасного суспільства, саме тому протягом останніх років відділення психоневрології для дітей з перинатальною патологією та орфанними захворюваннями ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» активно працює на теренах вивчення даних нозологій. Термін «орфанне (або рідкісне) захворювання» є досить новим та визначає захворювання, що загрожує життю людини або хронічно прогресує, призводить до скорочення тривалості життя громадянина або до його інвалідності, поширеність якого серед населення не більше ніж 1 : 2000. Хоча вперше цей термін був введений у медичну номенклатуру в 1983 році, на сьогодні вже відомо близько 7000 орфанних захворювань [1]. Близько 80 % рідкісних захворювань є генетично обумовленими, і більшість із них мають несприятливий для життя прогноз [2]. Майже 75 % орфанних хвороб проявляються в ранньому дитячому віці, а 30 % пацієнтів помирає до 5-річного віку [3].
Особливий акцент ми хотіли би зробити на проблемі діагностики раритетних хвороб. Через те, що вони рідко зустрічаються в популяції, зазвичай до постановки діагнозу пацієнти відвідують не одного спеціаліста, а в 40 % випадків рідкісне захворювання неправильно діагностується як мінімум один раз. P.A. Engel et al. у своїх публікаціях звертають увагу на те, що середній час від перших симптомів орфанного захворювання до встановлення точного діагнозу становить близько 4,8 року [4]. У медичній спільноті необхідно більше уваги приділяти обізнаності лікарів із рідкісним захворюванням, оскільки дані хвороби досить часто ховаються під масками, тобто симптомами, схожими на прояви інших відомих та поширених хвороб, що нерідко вводить лікаря в оману та призводить до встановлення хибного діагнозу [5].
У даній публікації ми наводимо власний досвід діагностики фатального орфанного захворювання — хвороби Канаван, а також сучасні дані світової літератури стосовно даної патології. Особливістю цього захворювання є те, що воно перебігає в ранньому дитячому віці під масками таких станів, як гідроцефалія, макроцефалія або дитячий церебральний параліч, але має прогресуючий перебіг і летальний кінець. Отже, коли ми оглядаємо дитину з відомим на перший погляд синдромом, завжди необхідно намагатися виявити етіологічний фактор, що призвів до розвитку неврологічних порушень. Свого часу відомий український дитячий вчений-невролог С.К. Євтушенко писав стосовно ДЦП: «Только в постсоветских странах остался не международный термин — ДЦП, что утвердило данную патологию как болезнь, а не как синдром!!! В связи с чем замедлился поиск этиологии этого тяжелого заболевания, а все устремления направлены на паллиативную реабилитацию (включая реабилитационные центры, где этиология не уточняется). Поэтому возникает мысль: «Мы не знаем, что лечим» [6].
На сьогодні в медичній літературі хворобу Канаван віднесено до групи лейкодистрофій, тобто тяжких спадкових порушень метаболізму, з переважним ураженням білої речовини головного мозку. У закордонній літературі можна зустріти синоніми даного захворювання: лейкодистрофія Канаван — Ван Богарта — Бертранда, спонгіозна дегенерація центральної нервової системи (ЦНС), інфантильна спонгіозна дегенерація, дефіцит аспартоацилази та N-ацетиласпартатова ацидурія [7].
Уперше дане захворювання описали Globus та Strauss у 1928 році, а пізніше в 1931 Міртель Мей Мур Канаван (Myrtelle May Moore Canavan) описала випадок захворювання дитини з прогресуючим ростом голови та летальним кінцем, у якої при автопсії була виявлена спонгіозна дегенерація білої речовини головного мозку [8]. Міртель Канаван — американський патологоанатом, закінчила медичний факультет Мічиганського університету в 1905 році. У 1910 році стала патологоанатомом у Бостонській державній лікарні, а в 1924 році була призначена патологоанатомом відділення психічних захворювань Массачусетського госпіталю. Великий інтерес у неї викликало вивчення патології нервової системи, тому паралельно з основною роботою вона була викладачем невропатології в Університеті Вермонта [9]. У 1920 році Міртель Канаван була призначена директором лабораторії Бостонської психопатологічної лікарні. З 1920 року до 1945 року була професором невропатології Бостонського університету та куратором відомого анатомічного музею Уоррена в Гарвардській медичній школі, де вона поповнила колекцію 1500 новими препаратами. Померла Міртель Канаван у 1953 році від хвороби Паркінсона [10].
У 1949 році Ван-Богарт та Бертранд описали подібну картину макроцефалії зі спонгіозною дегенерацією ЦНС у дітей із популяції євреїв-ашкеназі [11]. Майже через півстоліття було виявлено етіологічний чинник хвороби Канаван: спочатку Matalon et al. у 1988 році виявили підвищений рівень N-ацетиласпартату сечі хворих дітей, а в 1993 році було виявлено ген ASPA, мутації якого викликають дане захворювання [12]. З широким упровадженням та розробкою методик магнітно-резонансної томографії (МРТ) на початку ХХІ століття стало можливим проведення МРТ-спектроскопії та виявлення підвищеного рівня N-ацетиласпарагінової кислоти (NАА) у структурі білої речовини головного мозку хворих дітей [13].
На сьогодні виділяють дві клінічні форми хвороби Канаван — неонатальну та ювенільну. Дана градація є не патогенетичною, а виключно клінічною, оскільки процес ураження нервової системи починається в перинатальному періоді [14]. У більшості випадків спостерігається саме неонатальна, або інфантильна, форма хвороби Канаван. Діти народжуються клінічно здоровими, після чого триває період благополуччя, однак у віці 3–5 місяців починає розвиватись макроцефалія й затримка психомоторного розвитку. Затримка розвитку прогресує з кожним наступним місяцем життя. У новонароджених порушується смоктання, фіксація погляду та контроль за утриманням голови. У подальшому в дітей з хворобою Канаван вповільнюється моторний розвиток — відсутні навики сидіння, вставання та ходьби, також приєднується затримка домовленнєвого розвитку у вигляді відсутності гуління та лепету. Відмічаються порушення соціальної взаємодії у вигляді відсутності фіксації погляду та посмішки, однак деякі діти можуть взаємодіяти з оточуючими, посміхатися та слідкувати за предметами. М’язовий тонус на початку захворювання знижений, однак із часом змінюється на спастичний. У хворих дітей розвивається атрофія зорового нерва, що зумовлює відсутність реакції стеження та фіксації погляду. Ураження слуху не характерне [13–15]. З віком симптоми захворювання прогресують, у термінальних стадіях пацієнти переважно не можуть рухатися та ковтати, розвиваються епілептичні напади. Прогноз при хворобі Канаван несприятливий, більшість пацієнтів не доживає до другого десятиріччя життя [16].
При проведенні нейросонографії в дітей з неонатальною формою хвороби Канаван може бути виявлено підвищення ехогенності білої речовини головного мозку [14]. У неонатальному періоді комп’ютерна томографія (КТ) та МРТ головного мозку не виявляють типові зміни білої речовини [17, 18]. На автопсії зазвичай відмічається субкортикальна спонгіозна дегенерація білої речовини. При електронній мікроскопії виявляються роздуті астроцити з пошкодженими мітохондріями [19].
Залежно від типу генетичної мутації гена ASPА можуть спостерігатися різні клінічні прояви хвороби Канаван. Діти з гомозиготною мутацією Tyr231Ter (з відсутністю ферментної активності) клінічно відрізняються від осіб з гомозиготною Glu285Ala мутацією (залишкова ферментна активність) відсутністю різкого прогресування захворювання. Як правило, у неєвреїв у більшості випадків виявляється гомозиготний варіант Ala305Glu (без залишкової активності ферменту) на відміну від мутацій у євреїв ашкеназі, у яких захворювання може супроводжуватися легкою розумовою відсталістю [19].
Діти з ювенільною формою хвороби Канаван, як правило, народжуються без ознак ураження, у грудному періоді ростуть та розвиваються з мінімальною затримкою психомоторних навиків. При обстеженні таких дітей часто виявляється підвищений рівень N-ацетиласпартату в сечі. У більшості випадків діти з даною формою захворювання відвідують початкову школу за наявності належного догляду та реабілітації. На МРТ головного мозку при даній формі захворювання можуть бути виявлені зміни у вигляді підвищеного сигналу в базальних гангліях. Для ювенільної форми хвороби Канаван характерні одиничні мутації (Tyr288Cys, Arg71His або Pro257Arg) із залишковою активністю ферменту гена ASPA, що зумовлює «м’який» перебіг захворювання. У таких випадках може бути виявлений підвищений рівень ацетиласпартату в сечі та відсутність змін на МРТ головного мозку [18, 20, 21].
Хвороба Канаван — класична форма генетично детермінованої лейкодистрофії. Мутація виникає в гені — регуляторі ферменту аспартоацилази (ASPA), що бере участь у метаболізмі N-ацетиласпартату. Дана мутація має автосомно-рецесивний тип успадкування. Клінічні прояви хвороби Канаван виникають при успадкуванні аномального гена від кожного з батьків і прогностично є менделюючим захворюванням. Якщо людина отримує один нормальний ген і один мутантний ген, вона буде носієм цієї хвороби, але, як правило, клінічні прояви відсутні.
Ген хвороби Канаван був картований на 17-й хромосомі (17pter-P13). Даний ген ASPА кодує аспартоацилазу — фермент, що метаболізує N-ацетиласпарагінову кислоту. N-ацетиласпарагінова кислота є сполукою, що відіграє важливу роль у функціонуванні білої речовини мозку. У результаті виникнення дефіциту аспартоацилази відбувається накопичення N-ацетиласпарагінової кислоти в тканині головного мозку. Ознакою хвороби Канавана є аномально високий рівень N-ацетиласпарагінової кислоти, що може бути виявлений при проведенні магнітно-резонансної спектроскопії. Одна з теорій патогенезу відводить важливу роль N-ацетиласпарагіновій кислоті як компоненту регуляції водного метаболізму в білій речовині головного мозку. При порушенні функції аспартоацилази виникає хронічний набряк з утворенням кістоподібних порожнин (спонгіозна вакуолізація) у білій речовині мозку, а також утворюються аномальні мітохондрії у астроцитах [13, 17].
Хвороба Канаван не має статевої вибірковості, частота ураження осіб чоловічої та жіночої статі однакова. Виявлена висока частота носійства мутантного гена в популяції євреїв-ашкеназі (близько 1 на 40–58 осіб). Ризик народження хворої дитини серед євреїв-ашкеназі становить від 1 : 6 400 до 1 : 13 456. Загальна поширеність та захворюваність на сьогодні не відомі [22].
Діагностика в осіб із неонатальною формою хвороби Канаван спирається на показники високої концентрації N-ацетиласпарагінової кислоти в сечі. При ювенільній формі хвороби Канаван показник N-ацетиласпарагінової кислоти сечі може бути злегка підвищений. Зокрема, у дослідженнях (Michals& Matalon, 2011) було виявлено, що при неонатальній формі рівень NAA у сечі був підвищений у 62 рази, тоді як при ювенільній формі відзначалось підвищення всього в 4,6 раза порівняно з групою здорових дітей. Отже, аналіз сечі є відносно дешевим та інформативним методом для скринінгової діагностики даного захворювання. Остаточно для підтвердження діагнозу необхідна молекулярно-генетична діагностика мутації гена ASPA, що кодує фермент аспартоацилазу [21, 23]. Також проводиться визначення активності аспартоацилази в культурі фібробластів, причому зниження її активності підтверджує діагноз навіть за відсутності мутації гена ASPA. Електроенцефалограма може бути корисною у виявленні епілептиформної активності, при цьому фонова активність зазвичай є дифузно уповільненою [16].
Пренатальна генетична діагностика для групи ризику вагітності може бути здійснена шляхом вимірювання концентрації N-ацетиласпарагінової кислоти в амніотичній рідині [24]. Проте остаточний діагноз виставляється після проведення молекулярно-генетичного дослідження.
МРТ дозволяє виявити дифузну дегенерацію білої речовини переважно в субкортикальних відділах. У той же час центральні ділянки білої речовини, такі як перивентрикулярна зона та внутрішня капсула, часто залишаються інтактними на початку захворювання. У подальшому в патологічний процес залучаються бліда куля та таламус, однак хвостате ядро та лушпина не вражаються [25].
На сьогодні ефективного патогенетичного та етіологічного лікування немає, використовується тільки симптоматична терапія. Проте проводяться клінічні випробовування нових методів лікування, а саме: описані методики втручання в генетичну поломку гена ASPА для усунення дефектного гена [26]. Інший напрямок — це метаболічна терапія з використанням цитрату літію, що має здатність зменшувати рівень N-ацетиласпарагінової кислоти в головному мозку [27]. Також виявлено здатність гліцерилу триацетату зменшувати рівень N-ацетиласпарагінової кислоти в мозку хворих мишей та покращувати моторні функції. На сьогодні терапія гліцерилу триацетатом (у дозі до 4,5 г/кг) використовується при лікуванні дітей, у тому числі новонароджених, із хворобою Канаван [28]. Серед інших метаболічних засобів, ефективність яких на даний момент досліджується, слід назвати тригептатоїн, здатний знижувати оксидантний стрес, збільшувати кількість мієліну в мозку [29].
Переважно застосовується симптоматична терапія у вигляді антиконвульсантів при епілептичних нападах або сечогінних засобів при підвищенні внутрішньочерепного тиску [16]. Спираючись на власний досвід, ми рекомендуємо застосування в комплексному лікуванні метаболічної терапії з антиоксидантною та енерготропною дією. Застосовується коензим Q10 (4 мг/кг/добу), вітамін Е (200–400 мг /добу), фолієва кислота (0,5–2,5 мг/кг/добу), L-аргінін (500 мг/кг/добу внутрішньовенно краплинно), тіамін і рибофлавін (по 50 мг/добу), L-карнітин 100 мг/кг/добу. Важливим є також індивідуальний підбір антиконвульсивної, а при необхідності — психотропної терапії.
Наводимо опис власного клінічного спостереження дитини з хворобою Канаван, що запам’яталося нам складним діагностичним шляхом від початкових проявів захворювання до постановки остаточного діагнозу. Хлопчик М., 1 р. 8 міс., надійшов у відділення психоневрології нашого інституту в травні 2016 р. зі скаргами матері на наявність у дитини затримки статокінетичного й передмовленнєвого розвитку, порушень ковтання та епілептичних нападів у анамнезі.
Дитина народилася від І вагітності, що перебігала на фоні загрози передчасного переривання на 7-му та 16-му тижнях гестації, мати отримувала лікування. Зі слів матері, при проведенні УЗД на 23, 36 та 40-му тижнях патологічних змін не було виявлено. Пологи термінові, шляхом екстреного кесарського розтину у зв’язку зі слабкістю пологової діяльності. Маса при народженні 3560 г, довжина тіла 55 см, окружність голови 37 см, оцінка за шкалою Апгар 7/7 балів. На 3-й день дитина була виписана з пологового будинку. Вакцинопрофілактика не проводилася.
Перші прояви хвороби виникли у віці 1 міс. У дитини виникли порушення ковтання, підвищена збудливість. Отримувала лікування в районній лікарні за місцем проживання з діагнозом: синдром м’язового гіпертонусу, афект-респіраторний синдром унаслідок гіпоксично-ішемічного ураження ЦНС. При проведенні нейросонографії (НСГ) виявлено розширення передніх рогів бічних шлуночків, розширення судинних сплетень на рівні каудоталамічної вирізки. Діагностовано страбізм унаслідок парезу окорухового нерва справа. У 5 місяців у хлопчика було встановлено затримку розвитку: не утримував голову, не перевертався, не брав іграшки, не фіксував погляд. Проведена аксіальна комп’ютерна томографія: виявлено ознаки виражених дифузних енцефалопатичних змін, дифузної кортикальної атрофії з переважним ураженням лобної та скроневої частин мозку, помірне симетричне розширення елементів шлуночкової системи мозку. Встановлено діагноз змішаної гідроцефалії, затримки передмовленнєвого та статокінетичного розвитку, псевдобульбарного синдрому. У 8 місяців під час гіпертермії з’явилися клоніко-тонічні судоми. За даними електроенцефалографії (ЕЕГ) виявлено дифузні зміни біоелектричної активності мозку на фоні підвищеної збудливості. В 11 місяців встановлено діагноз: дитячий церебральний параліч, спастичний тетрапарез, субкомпенсована внутрішня та зовнішня гідроцефалія, псевдобульбарний синдром, симптоматична епілепсія з генералізованими тоніко-клонічними нападами (на фоні терапії вальпроатом натрію, адренокортикотропним гормоном напади припинились).
У квітні 2016 року (1 р. 7 міс.) у дитини виник генералізований тоніко-клонічний напад. Дитина була госпіталізована в неврологічне відділення за місцем проживання, після лікування в якому для уточнення діагнозу та корекції терапії була направлена в ДУ «ІПАГ НАМН України».
Неврологічний статус при надходженні: дитина у свідомості, реагує на звернену мову, посміхається. Експресивна мова відсутня. Відмічається вимушена поза — голову не утримує, верхні кінцівки у флексорному гіпертонусі, нижні кінцівки в екстензорному гіпертонусі, не перевертається, самостійне жування відсутнє, ковтає, однак захлинається рідиною. Голова гідроцефальної форми, окружність 52 см (> 3 SD), велике тім’ячко — 0,5 × 0,5 см, ненапружене, відмічається напруженість потиличних м’язів. Очні щілини симетричні, зіничні рефлекси ослаблені, симетричні. Страбізм справа внаслідок парезу окорухового нерва. За предметом не слідкує. Обличчя симетричне, міміка присутня, відмічається високий рефлекс із м’якого піднебіння, рухи голови обмежені. Тонус м’язів у верхніх та нижніх кінцівках підвищений за пірамідним типом, сухожилкові рефлекси з розширенням рефлексогенних зон, пожвавлені, з елементами клонусу, D = S. Тактильна й больова чутливість збережена. При підтримці — опора на передню частину стоп. Крокові рухи не виконує. Функції тазових органів не контролює. У соматичному статусі — патології не виявлено.
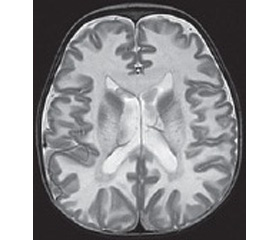
Проведено обстеження: загальний та біохімічний аналізи крові, загальний аналіз сечі — без патологічних змін. Електрокардіограма — синусовий ритм, помірні обмінні зміни в міокарді. ЕЕГ — виражені дифузні неспецифічні зміни біоелектричної активності головного мозку, уповільнення фонової активності, пароксизмальної епілептичної активності не виявлено. УЗД органів черевної порожнини — ультразвукові ознаки помірних реактивних змін паренхіми печінки. НСГ — підвищення ехогенності мозкової паренхіми, вентрикулодилатація ІІ ст. За даними МРТ головного мозку виявлено підвищену інтенсивність сигналу від білої речовини, розширення шлуночкової системи, ймовірно, обумовлені захворюванням із групи генетично детермінованих токсичних і метаболічних енцефалопатій (рис. 1). Консультація офтальмолога: виявлено пеленчату атрофію дисків зорових нервів. Консультативний висновок медичного генетика: у дитини не виключається спадкове дегенеративне захворювання ЦНС: лейкодистрофія Канаван, автосомно-рецесивний тип успадкування. Проведено визначення каріотипу: 46,ХY — нормальний чоловічий каріотип. При молекулярно-генетичному дослідженні ДНК, виділеної з крові пацієнта, у гені ASPА виявлена мутація р.Ala305Glu, що дало можливість остаточно підтвердити діагноз запідозреної хвороби. У сім’ї було проведене генетичне консультування з рекомендацією щодо проведення обов’язкової пренатальної діагностики при наступній вагітності.
/49-1.jpg )
За час перебування у відділенні дитина отримувала симптоматичну (вальпроат натрію — 30 мг/кг), протинабрякову (магнію сульфат, L-лізину есцинат), міорелаксуючу (баклофен) та ноотропну терапію (цитиколін). Відмічалося покращення стану дитини у вигляді припинення епілептичних нападів та зниження м’язового тонусу. Після встановлення остаточного діагнозу повторно батьки по допомогу не зверталися.
Висновки
Лейкодистрофії, до яких належить описана вище хвороба Канаван, є одними з найбільш некерованих прогредієнтних захворювань, описаних у дитячій неврології. Встановлення в дитини даного діагнозу, безумовно, є тяжкою подією не тільки для родини, але й для медичних працівників, які опікуються нею. Рівень розвитку сучасної медицини, на превеликий жаль, не дозволяє відносити дане захворювання до курабельних. Неухильно прогресуючий перебіг, з ураженням основних функцій нервової системи, з часом призводить до фатальних наслідків. Завданням лікаря є перш за все запідозрити прогресуюче захворювання в дитини з нетиповою картиною органічного ураження нервової системи та своєчасно розпочати діагностичний пошук у даному напрямку. Встановлення адекватного діагнозу необхідне для родини, щоб позбавити її марного витрачання часу в пошуках вірного діагнозу та проведення необґрунтованого дорогого лікування, а ресурси направити на догляд за дитиною та проведення симптоматичного лікування, що може надовго подовжити її життя. Вважаємо за доцільне звернути увагу на те, що нашому пацієнту необхідно було б провести магнітно-резонансну спектроскопію для визначення рівня N-ацетиласпартату в речовині головного мозку, але це дослідження не було проведене у зв’язку з матеріальною неспроможністю батьків (дане дослідження проводиться в приватних медичних центрах). Хотілося б акцентувати увагу на необхідність створення простору для обміну досвідом між лікарями та родинами пацієнтів, активне їх залучення до обговорення проблем дітей з орфанними захворюваннями на державному рівні.