Резюме
Мета роботи: аналіз особливостей клінічної картини хвороби Помпе (ХП), визначення за результатами клініко-лабораторних і інструментальних досліджень основних характеристик міопатичних синдромів, що дозволяють виділити таких хворих у групу селективного скринінгу для проведення ферментної діагностики на наявність ХП. Матеріали та методи. У період з 2002 по 2016 р. у Центрі орфанних захворювань обстежені 614 хворих з клінічними проявами спадкового нервово-м’язового захворювання. Вік пацієнтів — від 2 міс. до 34 років; хворих чоловічої статі — 314, жіночої — 300. Результати. У 78 хворих виявлена міопатія домінантного типу успадкування, подальше лабораторне дослідження на ХП у них не проводили. У 98 пацієнтів діагностована спінальна м’язова атрофія, у 70 — первинна м’язова дистрофія Дюшена, у 123 — різні форми спадкової нейросенсорної невропатії. Скринінгова група з метою виявлення ХП методом ферментодіагностики — 421 хворий, у них спостерігали некласифіковані міопатичні синдроми, що характеризувалися гетерогенною клінічною картиною, зокрема загальною слабкістю м’язів, відсутністю їх гіпертрофії. Електронейроміографія виконана в 187 пацієнтів: у всіх виявлені патерни міопатичного процесу без ознак денервації, спонтанної активності у вигляді фібриляцій або міотонічних феноменів. У 25 хворих віком від 2 до 18 міс. виявлений підвищений рівень креатинфосфокінази, лактатдегідрогенази, печінкових трансаміназ. Крім того, у 10 хворих діагностовані гіпертрофічна кардіоміопатія, розлади дихання. За результатами ферментодіагностики в сухій краплі крові у 5 хворих діагностована ХП. Висновки. Визначені основні характеристики групи селективного пошуку, у якій діагностика ХП методом ферментодіагностики в сухій краплі крові є ефективною. Такими характеристиками є: автосомно-рецесивний тип успадкування захворювання; широкі вікові інтервали; підвищення рівня креатинфосфокінази, печінкових трансаміназ; локалізація міопатичного ураження (переважно м’язи проксимальних відділів кінцівок і тулуба), характерні зміни за даними електронейроміографії, гіпертрофічна кардіоміопатія.
Цель работы: анализ особенностей клинической картины болезни Помпе (БП), определение по результатам клинико-лабораторных и инструментальных исследований основных характеристик миопатических синдромов, позволяющих выделить таких больных в группу селективного скрининга для проведения ферментодиагностики на наличие БП. Материалы и методы. В период с 2002 по 2016 г. в Центре орфанных заболеваний проведено комплексное клинико-лабораторное исследование у 614 больных с клиническими проявлениями наследственного нервно-мышечного заболевания. Возраст пациентов — от 2 мес. до 34 лет; больных мужского пола — 314, женского — 300. Результаты. У 78 больных выявлена миопатия доминантного типа наследования, в дальнейшем лабораторные исследования на БП у них не проводили. У 98 пациентов диагностирована спинальная мышечная атрофия, у 70 — первичная мышечная дистрофия Дюшена, у 123 — различные формы наследственной нейросенсорной невропатии. Скрининговая группа для выявления БП методом ферментодиагностики — 421 больной, у которого наблюдали неклассифицированные миопатические синдромы, характеризовавшиеся гетерогенной клинической картиной, в частности общей слабостью мышц, отсутствием их гипертрофии. Электронейромиография выполнена у 187 пациентов: у всех выявлены паттерны миопатического процесса без признаков денервации, спонтанной активности в виде фибрилляций или миотонических феноменов. У 25 больных в возрасте от 2 до 18 мес. выявлен повышенный уровень креатинфосфокиназы, лактатдегидрогеназы, печеночных трансаминаз. Кроме того, у 10 больных диагностированы гипертрофическая кардиомиопатия, расстройства дыхания. По результатам ферментодиагностики в сухой капле крови у 5 больных диагностирована БП. Выводы. Определены основные характеристики группы селективного поиска, в которой диагностика БП методом ферментодиагностики в сухой капле крови является эффективной. Такими характеристиками являются: аутосомно-рецессивный тип наследования заболевания; широкие возрастные интервалы; повышение уровня креатинфосфокиназы, печеночных трансаминаз; локализация миопатического поражения (преимущественно мышцы проксимальных отделов конечностей и туловища), характерные изменения по данным электронейромиографии; гипертрофическая кардиомиопатия.
Background. Analysis of Pompe disease (PD) clinical features, determination of myopathic syndromes main characteristics according to the results of clinical, laboratory and instrumental studies allowing to select such patients into screening group for PD enzyme diagnosis. Materials and methods. In 2002–2016, in the Center of orphan diseases a comprehensive clinical and laboratory study was conducted in 614 patients with clinical manifestations of hereditary neuromuscular disease. There were 314 male and 300 female patients aged 2 months to 34 years. Results. In 78 patients, myopathy with dominant inheritance was detected, further laboratory tests on PD haven’t been conducted. In 98 patients, spinal muscular atrophy was diagnosed, in 70 — Duchenne primary muscular dystrophy, in 123 — different forms of hereditary neurosensory neuropathy. Screening group for PD detection using enzyme diagnosis method consisted of 421 patients, who had unclassified myopathic syndromes characterized by a heterogeneous clinical picture, in particular, general muscle weakness without hypertrophy. Electroneuromyography was performed in 187 patients: all of them showed patterns of myopathic process without signs of denervation and spontaneous activity (fibrillation or myotonic phenomena). In 25 patients aged 2 to 18 months, increased levels of creatine phosphokinase, lactate dehydrogenase, and liver transaminases were detected. Moreover, in 10 patients, hypertrophic cardiomyopathy, respiratory distress were revealed. According to enzyme diagnosis results, PD in dried blood spot was diagnosed in 5 patients. Conclusions. Main characteristics for the selective search group on PD were determined, in which enzyme diagnosis in dried blood spot was effective. Such characteristics are: autosomal recessive type of disease inheritance; wide age intervals; increased levels of creatine phosphokinase, hepatic transaminases; myopathic lesion localization (predominantly in muscles of proximal parts of the extremities and body), characteristic electroneuromyography changes; hypertrophic cardiomyopathy.
Вступ
Нервово-м’язові захворювання посідають перше місце за частотою серед усіх спадкових моногенних неврологічних захворювань; їх виявляють із частотою 1 на 3500 населення [1]. Спадкові нервово-м’язові захворювання — клінічно та генетично гетерогенна група захворювань, які класифікують за типом успадкування, локалізацією патогенетичного процесу, зокрема міо–патії, типом мутантного білка. Переважна більшість захворювань цієї групи характеризується невпинним прогресуючим перебігом, ефективні методи лікування відсутні [2].
Проте останнім часом досягнуті суттєві успіхи не тільки в діагностиці, а й у лікуванні окремих типів нервово-м’язових захворювань, зокрема з групи спадкових метаболічних міопатій [3].
Із 2006 р. стало можливим лікування однієї з метаболічних міопатій — дефіциту кислої альфа-глюкозидази (GAA; кислої мальтази), або хвороби Помпе (ХП), з використанням препарату альфа-глюкозидази [4].
ХП — рідкісне (орфанне) нервово-м’язове захворювання з прогредієнтним перебігом (OMIM232300), яке належить до лізосомальних хвороб накопичення та має автосомно-рецесивний тип успадкування. Ген, що кодує GAA, складається з 20 екзонів і картований на хромосомі 17q25.2-q25.3. На сьогодні описані 360 мутацій, що спричиняють часткове або цілковите блокування каталітичної активності ферменту, зменшення його стабільності та періоду напіврозпаду.
За характером накопичуваного субстрату ХП належить до глікогенозів II типу. В основі патогенезу ХП лежить зниження активності (або цілковита відсутність) ферменту лізосом — кислої альфа-глюкозидази, що бере участь у розщепленні глікогену. Через низьку активність GAA нерозщеплений глікоген накопичується в лізосомах клітин різних тканин, переважно в поперечносмугастих м’язах тулуба, кінцівок і міокарда, спричиняючи порушення функції та структури м’язових волокон, що призводить до механічної руйнації та загибелі міоцитів [5].
За даними американських і європейських дослідників, ХП становить близько 15 % у структурі всіх хвороб накопичення глікогену, які на даний час розподіляють на 14 типів [6].
Частота виявлення ХП становить 1 випадок на 40 000 живонароджених. Проте, за даними досліджень останніх років, поширеність ХП суттєво варіює в різних популяціях залежно від етнічної приналежності: від 1 : 14 000 до 1 : 300 000 живонароджених [7].
Клінічна картина ХП характеризується широким спектром симптомів і варіантів перебігу [8]. Дотепер детально описана лише одна, класична форма ХП — інфантильна (ІФХП), яку вперше описав у 1932 р. J.C. Pompe [9]. В основу найбільш поширеної класифікації ХП покладено вік хворого, у якому виникли перші симптоми захворювання; виділяють ІФХП, або ХП з раннім початком (ХПРП), і ХП з пізнім початком (ХППП) [5]. Існують два клінічних варіанти ХПРП: класичний інфантильний і атиповий (некласичний). Класична ІФХП — фатальна прогресуюча міопатія з генералізованою м’язовою слабкістю, гіпотонією, гіпертрофічною кардіоміопатією. За відсутності лікування при класичній ІФХП у хворих першого року життя виникає серцево-легенева недостатність; тривалість життя таких пацієнтів становить у середньому 8 міс. Атипова ІФХП характеризується повільним перебігом, затримкою розвитку моторних функцій на тлі генералізованої м’язової слабкості, можливі незначні порушення з боку міокарда. За відсутності лікування хворі живуть у середньому до двох років. Активність GAA у фібробластах шкіри хворих при ХПРП не перевищує 1 % від такої в нормі [10].
При ХППП перші ознаки захворювання виникають у строки від кількох місяців з моменту народження до шістдесяти років життя. У хворих відзначають прогресуючу м’язову слабкість, зазвичай без суттєвих змін із боку серця; з часом виникає дихальна недостатність і потреба в штучній вентиляції легень. Саме дихальна недостатність спричиняє ранню смерть таких пацієнтів [11]. Також описані спостереження ранньої смерті таких хворих унаслідок розриву аневризм базилярної артерії [12]. Активність GAA у фібробластах шкіри пацієнтів при ХППП становить 1–40 % від норми.
Золотим стандартом лабораторної діагностики ХП є визначення активності GAA у лейкоцитах або культурі фібробластів [13]. Крім того, останніми роками розроблений і широко застосовується на практиці метод визначення активності GAA у сухій плямі крові (СПК). Проте цих програм недостатньо для вирішення проблем діагностики ХП, особливо якщо перші ознаки захворювання виникають у дорослому віці й воно має м’який перебіг [14].
При ХП існує кореляція «генотип – фенотип», тому важливим є молекулярний аналіз гена GAA. Для встановлення точного діагнозу ХП достатньо визначення активності GAA.
Рання клінічна та лабораторна діагностика ХП, призначення патогенетичної ферментозамісної терапії (ФЗТ) як результат проведення універсального скринінгу сприяли б суттєвому підвищенню ефективності лікування та запобігали б ранній смерті таких хворих. Затримка діагностики, особливо при ХППП, — досить звичне явище, інколи діагноз встановлюють майже через десять років з моменту виникнення перших симптомів захворювання [15]. Пілотні програми неонатального скринінгу, розробка яких триває (а подекуди (США, Тайвань) вони вже впроваджені в клінічну практику), сприятимуть своєчасній діагностиці ХП [16]. Проте програми масового неонатального скринінгу не вирішують питання діагностики ХП у вже наявних хворих різного віку, які залишаться поза межами масового неонатального скринінгу.
Саме тому розробка ефективних діагностичних алгоритмів є найважливішим етапом організації та надання медичної допомоги пацієнтам при ХП.
Мета роботи — аналіз особливостей клінічного перебігу ХП; визначення клініко-лабораторних характеристик пацієнтів зі міопатичними синдромами, яких можна включити в групу селективного скринінгу для проведення ферментної діагностики на ХП.
Матеріали та методи
Упродовж 2002–2016 рр. у Центрі орфанних захворювань НДСЛ «Охматдит» обстежені 614 хворих (314 — чоловічої статі, 300 — жіночої) з клінічними проявами спадкового нервово-м’язового захворювання. Вік хворих — від 3 міс. до 34 років; вік 522 (85 %) пацієнтів — від 3 міс. до 15 років.
Усім хворим проведені комплексне клініко-лабораторне та інструментальне обстеження, генеалогічний аналіз. Обов’язково визначали рівень креатинфосфокінази (КФК), печінкових трансаміназ (аланінамінотрансферази (АЛТ), аспартатамінотрансферази (АСТ)), лактатдегідрогенази (ЛДГ). У 464 (75,6 %) хворих виконана електронейроміографія (ЕНМГ), у 350 (57 %) — електрокардіографія і ехокардіографія (ЕхоКГ). За наявності показань (згідно з даними клініко-лабораторного дослідження) проводили молекулярно-генетичне дослідження мутацій у гені SMN і гені дистрофіну (Інститут молекулярної біології та генетики НАН України); молекулярно-генетичне дослідження мутацій у гені GAA на ХП (лабораторія Archimed Life Science GmbH, Відень, Австрія); визначення активності GAA у СПК (медико-генетична лабораторія НДСЛ «Охматдит») за модифікованим протоколом N.A. Chamоles і спів–авт. [17] із використанням плашечного аналізатора Wallac VіctоrTM 1420 Multіlabel Cоunter (Perkіn Elmer, Турку, Фінляндія).
Результати та обговорення
За результатами генеалогічного аналізу в 78 (12,7 %) пацієнтів виявлений домінантний тип успадкування захворювання, у подальшому лабораторні дослідження з метою підтвердження діагнозу ХП у них не проводили. У 98 (15,7 %) хворих за даними ЕНМГ і на підставі виявлених мутацій у гені SMN діагностована спінальна м’язова атрофія (СМА); у 70 (11,4 %) — первинна м’язова дистрофія Дюшена; у 123 (20 %) — різні форми спадкової нейромотосенсорної невропатії.
У 421 (68,6 %) хворого з гетерогенною клінічною картиною захворювання, м’язовою слабкістю виявлені некласифіковані міопатичні синдроми — цю групу пацієнтів вважали селективною (скринінговою) для виявлення ХП методом СПК. ЕНМГ виконана в 187 пацієнтів скринингової групи: в усіх спостереженнях виявлені патерни міопатичного процесу без ознак денервації, спонтанної активності у вигляді фібриляцій або міотонічних феноменів. У 25 хворих віком від 2 до 18 міс. відзначали підвищення рівня АЛТ і АСТ, креатинфосфокінази (вище від 220 од/л), лактатдегідрогенази (вище від 250 од/л). У 10 хворих спостерігали гіпертрофічну кардіоміопатію, дихальні розлади.
За результатами ферментодіагностики діагноз ХП встановлений у 5 пацієнтів: у 3 — ІФХП, у 2 — ХППП. Клінічна картина була досить гетерогенною, проте в усіх спостереженнях відзначали сповільнення збільшення маси тіла, затримку формування статокінетичних навичок.
Усі пацієнти народилися від нормальних вагітностей та фізіологічних пологів. Антропометричні показники при народженні — у межах популяційної норми.
Перші симптоми класичної форми захворювання (ІФХП) виникали в перші 1–3 міс. життя, атипової (ХППП) — від 6 міс. до 6 років (табл. 1).
У усіх спостереженнях діагноз встановлений із затримкою: при ІФХП — у віці хворого в середньому 5,1 міс., при ХППП — 6,5 року, що є характерним для цієї патології [18].
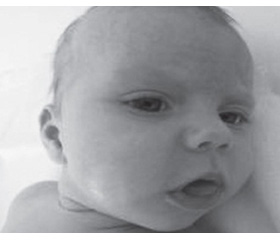
Основним у клінічній картині захворювання був міо–патичний синдром, вираженість якого різнилася залежно від форми ХП і віку пацієнтів. У хворих віком до 1 року спостерігали виражений міопатичний синдром: генералізовану м’язову слабкість (млява дитина), під час тракційного тесту — характерне закидання голови і симптомом «провислої білизни» (рис. 1).
У дітей із ХППП на тлі загальної гіпотрофії м’язового корсету відзначали виражену слабкість, переважно у проксимальних групах м’язів тулуба і кінцівок, сколіотичне порушення постави, деформацію грудної клітини («серцевий горб»). За даними існуючих у світі реєстрів хворих на ХП, скелетні деформації виявляють майже в 50 % таких хворих [19].
У хворих при ІФХП відзначали патогномонічний симптом — макроглосію (рис. 2); при ХППП вона відсутня. У 4 з 5 хворих виявлений птоз без порушення функції окорухових нервів. За даними літератури, відомості щодо наявності птозу в пацієнтів із ХП різняться: від відсутності цього симптому до його наявності у 23 % випадків у досліджуваних когортах пацієнтів із ХП [20, 21]. На нашу думку, очні симптоми, зокрема птоз, є надзвичайно важливими при диференціальній діагностиці ХП з іншими метаболічними міопатіями, наприклад мітохондріальними захворюваннями (синдром Лі, дефіцит комплексів дихального ланцюга мітохондрій тощо) [22].
/115-2.jpg)
Для хворих на ІФХП характерне виникнення порушень із боку серця. За даними ЕхоКГ, у всіх пацієнтів через 1–2 міс. з моменту виникнення перших ознак захворювання відзначали гіпертрофію лівого шлуночка, дилатаційну необструктивну кардіоміопатію. В усіх хворих при ІФХП на момент встановлення діагнозу відзначали виражений міопатичний синдром у поєднанні з прогресуючою гіпертрофічною кардіоміопатією, що спричиняла загрозливе зменшення фракції викиду крові з лівого шлуночка (від 67 до 45 %). Слід зауважити, що інколи ознаки гіпертрофії лівого шлуночка серця вдається діагностувати в пренатальному періоді [21].
Порушення функції серця також відзначали у двох хворих на ХППП, проте без прогресуючої гіпертрофії лівого шлуночка; крім того, в одного з цих пацієнтів виявлена рабдоміома лівих відділів серця.
У всіх хворих спостерігали порушення з боку шлунково-кишкового тракту: діарею, сповільнене збільшення маси тіла або її втрату. У старших дітей відзначали схильність до частих гострих респіраторних вірусних інфекцій (ГРВІ), які ускладнювалися виникненням дихальної недостатності та пневмонії. У 2 хворих через гострі порушення дихання встановлено трахеостому, ще один пацієнт потребував стаціонарної апаратної підтримки під час нічного сну. За даними літератури, прогресування ХП спричиняє фіброз легень, що зумовлює необхідність використання додаткових інструментальних методів моніторингу функції дихання в протоколі наданні медичної допомоги таким хворим (пульсоксиметрія, полісомнографія, моніторинг функції зовнішнього дихання) [23].
Виражену клінічну картину захворювання спостерігали у двох хворих віком 6 і 7 років. В одному спостереження ГРВІ спричинила виникнення спочатку гострих, а в подальшому — і хронічних розладів дихання, що суттєво погіршило стан хворого, у якого у віці 6 міс. виявлені ознаки міопатії. Через зазначені особливості перебігу захворювання пацієнт на 2 роки практично втратив здатність ходити самостійно. В іншому клінічному спостереженні вираженість міопатичного синдрому була мінімальною, проте він почав прогресувати на тлі перенесеного інтеркурентного захворювання, коли у хворого виникли гострі розлади дихання, а в подальшому — слабкість м’язів тулуба, проксимальних відділів кінцівок, формування специфічної ходи. Обидва пацієнти потребували встановлення трахеостоми та реанімаційної підтримки. В одного з них виникла вторинна кардіоміопатія легкого ступеня, прояви якої вдалося усунути за два роки безперервної ФЗТ.
За результатами лабораторних досліджень у всіх 5 хворих відзначали стабільно високі рівні КФК, ЛДГ, АЛТ, АСТ. КФК є неспецифічним маркером міопатичних процесів різної етіології: запальних, спадкових (зокрема, дистрофінопатій) і метаболічних (ХП, синдроми порушення мітохондріального бета-окислення тощо). Рівень КФК при ХП не перевищує 2000 од/л, однак у деяких хворих (особливо при ХППП, ознаки якої виникли в дорослому віці) цей показник може бути в межах норми [24]. У нашому дослідженні рівень КФК не перевищував 1500 од/л. Кореляція між рівнем КФК, формою захворювання та ступенем його тяжкості відсутня [7].
У всіх хворих відзначали зниження остаточної активності GAА, що не перевищувала 0,23 нмоль/год/мг білка. Рівень активності GAA корелює з тяжкістю захворювання. Так, у хворого за цілковитої відсутності активності GAA перебіг ХП був найтяжчим, дитина померла у віці 4 міс. Стан хворого з найвищим рівнем GAA був тяжким, проте клінічний перебіг захворювання характеризувався найбільш пізнім дебютом, збереженням щоденної рухової активності, здатності самостійно ходити. Хворому встановлена трахеостома, проте він не потребував нічної апаратної підтримки функції дихання. Кореляцію між рівнем остаточної активності GAA, формою й ступенем тяжкості ХП пояснюють генно-фенотиповими особливостями. Отримані нами результати збігаються з даними інших дослідників [21]. Хоча, за даними деяких авторів, пряма кореляція між рівнем остаточної активності GAA і тяжкістю ХП відсутня [19].
/116-1.jpg)
При ХП час встановлення діагнозу є критичним для початку ФЗТ, що запобігає виникненню необоротного ушкодження м’язів і, відповідно, життєво важливих органів і систем.
У 3 хворих на ХФ упродовж 3 років застосовували ФЗТ. Так, своєчасне призначення ФЗТ при ІФХП (клінічне спостереження 3) дозволило уникнути ранньої смерті дитини (ще двоє хворих на ІФХП, яким ФЗП не призначали, померли у віці до 7 міс.). З іншого боку, у 2 хворих на ХППП діагноз встановлений запізно, ФЗТ довелося розпочинати в реанімаційному відділенні. Однак навіть пізній початок ФЗТ дозволив зберегти життя хворих, відновити рухову активність, в одного пацієнта майже повністю відновилася здатність ходити самостійно, відзначене покращення функції дихальної та серцево-судинної систем. Невелика кількість спостережень не дозволяє зробити вірогідні висновки щодо ефективності ФЗТ, проте попередні результати нашого дослідження є досить оптимістичними й збігаються з даними інших дослідників [7].
566 (85 %) обстежених нами хворих були дитячого віку: у 3 них виявлена класична форма захворювання — ІФХП, у 2 — атипова, яку можна вважати проміжною між ІФХП і ХППП у дорослих осіб.
Аналіз частоти виявлення ХП у різних географічних та етнічних групах свідчить про те, що частіше це захворювання діагностують у дорослих осіб, у яких також описано асимптомний перебіг ХП [25, 26]. Саме тому дуже важливим є врахування вікових інтервалів селективного вибору пацієнтів для проведення подальшої лабораторної діагностики.
За результатами нашого дослідження й даними інших авторів [27], діагностика ХП є надзвичайно складною, у більшості спостережень діагноз встановлюють запізно. Клінічна картина ХП дуже варіабельна, подібна до більшості спадкових нервово-м’язових захворювань. Найбільш ефективною групою пошуку пацієнтів із ХП є група хворих із кінцівково-поперековими формами міопатії, у яких виявлена гіперкреатинфосфокіназемія [28].
Висновки
Клінічний дизайн селективного скринінгу на ХП із використанням методу СПК має включати широкий віковий інтервал, враховувати особливості локалізації міопатичного ураження, підвищення рівня КФК, печінкових трансаміназ, дані ЕНМГ, ЕхоКГ. Такий підхід дозволить підвищити ефективність діагностики та суттєво впливати на ефективність лікування пацієнтів із ХП.
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів при підготовці даної статті.

/114-1.jpg)
/115-1.jpg)