Кистозная болезнь почек — это группа наследственных или приобретенных заболеваний, которые характеризуются присутствием и ростом в почках единичных или множественных кист, представляющих собой закрытую полость, окаймленную эпителием и заполненную жидким или желеобразным экссудатом. По своей структуре киста представляет собой расширенный сегмент нефрона или собирательной трубки, а ее размер определяется количеством содержимого и варьирует от мелкого (менее 2 мм в диаметре) до гигантского. Кистозная жидкость сообщается с кровеносными сосудами, содержимым канальцев и почечной лоханки.
Почечные кисты классифицируют в зависимости от происхождения — негенетического и генетического (табл. 1).
Одной из форм кистозной болезни почек является поликистозная болезнь почек (ПБП). ПБП — это генетическое заболевание, которое приводит к образованию и прогрессированию множественных кист в паренхиме почек и в конечном итоге может привести к терминальной стадии болезни почек (ТПН). Это четвертая ведущая причина хронического заболевания почек, приводящая к ТПН.
Аутосомно-доминантная поликистозная болезнь почек (АДПБП) является наиболее распространенной формой ПБП с частотой 1 : 400 — 1 : 1000 случаев, которой страдают 12,5 млн человек во всем мире всех этнических групп и которая составляют 4–10 % больных ТПН, получающих лечение заместительной почечной терапией [1]. У 85–90 % пациентов АДПБП является результатом мутации гена PKD-1, в то время как 10–15 % случаев обусловлены мутацией гена PKD-2 [2]. Ген PKD-1 (polycystic kidney disease 1) кодирует интегральный мембранный белок полицистин-1 –(РС-1), а ген PKD-2 (polycystic kidney disease 2) — белок полицистин-2 (РС-2) [3].
Полицистин-1. Структура и функция
Белок РС-1 имеет молекулярную массу (м.м.) 462 кДа, состоит из 4302 аминокислот с 11 трансмембранными доменами. В своей структуре содержит большой внеклеточный N-конец и короткий С-конец [4]. РС-1 экспрессируется эпителиальными, развивающимися и зрелыми клетками почечных канальцев, а также различными тканями, включая сердце, печень, кости и эндокринные железы [5]. Самые высокие уровни РС-1 обнаружены в фетальной почечной ткани, а самые низкие — в тканях взрослых [6]. Полицистин-1 обнаружен в ресничках, локализуется в латеральном домене плазматической мембраны и адгезивном комплексе в поляризованных эпителиальных клетках. Большой внеклеточный N-конец РС-1 содержит 15 PKD повтора, два лейциннасыщенных повтора, окруженных цистеином, и лектиновый домен С-типа. Многие из этих доменов имеют решающее значение для функционирования РС-1 и играют существенную роль в белково-белковом или белково-матриксном взаимодействии. Это служит доказательством того, что комбинация субклеточной локализации РС-1 в плазматической мембране и соединительного комплекса подтверждает роль РС-1 во взаимодействиях «клетка — клетка», «клетка — матрикс». Внеклеточные домены РС-1 участвуют в процессах потока жидкости и давления в почках. С-терминальный конец РС-1, состоящий из 200 аминокислот, содержит G-связывающий белок и спирально-катушечный домен. С-терминальный конец содержит также последовательность, богатую пролином, глутаминовой кислотой, серином и треонином, которые могут способствовать его убиквитин-опосредованному снижению. Полицистин-1 подвергается расщеплению в N- и С-терминальных доменах. N-концевое расщепление происходит при участии G-белка на сайте протеолитического рецептора (GPS) непосредственно перед первым трансмембранным доменом [7] (рис. 1). Это цис-аутопротеолитический процесс расщепления, который происходит на ранней стадии секреторного пути, и расщепленный N-терминальный конец РС-1 остается нековалентно присоединенным к мембраносвязанному С-терминальному фрагменту. Не все РС-1 молекулы в клетке расщепляются, генерируя гетерогенную популяцию полноразмерных GPS-расщепленных РС-1-белков. Полностью функциональный РС-1 должен быть способен к N-концевому расщеплению. Экспрессия мутантной формы РС-1 не может осуществить расщепление GPS. Это проявление миссенс-мутации в случае аутосомно-доминантного поликистоза почек [8].
/17-1.jpg)
С-терминальный цитоплазматический остаток из РС-1 высвобождается путем двух расщеплений (рис. 2). При первом расщеплении высвобождается растворимая форма остатка (м.м. 35 кДа), которая аккумулируется в ядре в ответ на уменьшение потока жидкости в почках. Второе, дистальное, расщепление высвобождает фрагмент с м.м. 15 кДа из цитоплазматического остатка РС-1, который взаимодействует с активатором транскрипции STAT6 и коактиватором р100 [9]. При этом в клетках кист при АДПБП наблюдается усиление расщепления С-терминального остатка.
N-терминальный конец белка РС-1 расщепляется на протеолитическом сайте рецептора, связанного с белком (GPS). Высвобожденный С-концевой остаток перемещается в ядро вместе с компонентами Wnt, STAT6/p100 и, возможно, с другими регуляторами транскрипции.
Полицистин-2 и функционирование Са2+-каналов
Полицистин-2 представляет собой белок, имеющий 968 аминокислот, которые шесть раз пересекают мембрану с внутриклеточными N- и С-терминальными концами. РС-2 локализован в некоторых субклеточных компартментах. Большое его содержание обнаружено в эндоплазматическом ретикулуме и аппарате Гольджи [10]. Функциональный РС-2 обнаружен в плазматической мембране, где он может существовать в комплексе с РС-1. Пулы РС-2 также содержатся в большом количестве в субклеточных доменах, таких как реснички, и митотическом веретене [11].
Белок РС-2 функционирует как Са2+-прони–цаемый неселективный катионный канал и является гомологичным семейству катионных каналов с временным рецептором. Хотя часть РС-2 локализуется совместно с РС-1 в ресничке, основной клеточный пул РС-2 находится во внутриклеточных компартментах, где он может модулировать высвобождение кальция из внутриклеточного хранилища. Активность канала комплекса РС-1/РС-2 проявляется изгибом ресничек и может играть опосредованную роль ресничек в трансдукции других механических или химических раздражителей [12].
Несколько доменов, присутствующих в РС-2 на N- и С-терминальных остатках, являются ответственными за взаимодействие «белок — белок» и чувствительность к Са2+. Два домена, один из которых находится в цитоплазме, способствуют олигомеризации РС-2. Кальций-связывающий домен начинается вверху и распространяется до катушечно-спиральной области. Структура «спираль — петля — спираль» связывает Са2+, позволяя белку определить чувствительность для Са2+. РС-2 имеет единственный Са2+-связывающий сайт с микромолярной активностью. Мутация РС-2 способствует удалению этого С-терминального домена, изменяя все его взаимодействия и регулирования, что в итоге приводит к аутосомно-доминантному поликистозу почек (АДПКП) [13].
РС-2 является кальций-активированным каналом, что способствует высвобождению кальция из внутриклеточных хранилищ в ответ на локальное увеличение его концентрации. Кальциевый канал в РС-2 находится между 5-м и 6-м трансмембранными доменами, с некоторым вовлечением третьего домена [14]. Миссенс-мутация РС-2 приводит к нарушению функционирующего канала и является причиной АДПКП.
Необходимо отметить, что белок РС-2 не прямо регулирует уровень цитоплазматического Са2+, а осуществляет это через взаимодействие с рианодиновым рецептором и 1,4,5-трифосфат-инозитоловым рецептором [15].
Суммарная характеристика генов и белков при АДПКП представлена в табл. 2.
Взаимодействие между РС-1 и РС-2
Субклеточная локализация РС-1 и РС-2 позволяет им функционировать совместно [16]. Это наиболее выражено в первичной ресничке и в эндоплазматическом ретикулуме. Исследования, проведенные с использованием клеток из кист АДПКП, показывают, что нарушение функции одного белка отрицательно действует на локализацию другого. Так, мутация РС-1, приводящая к экспрессии клеток и связанная с АДПКП, предотвращает расщепление GPS и уменьшает количество РС-1 и РС-2 в первичных ресничках [17]. Взаимодействие между РС-1 и РС-2 подтверждает важное значение в функционировании Са2+-канала. Физически взаимодействие двух белков осуществляется через их С-терминальный цитоплазматический остаток [18] (рис. 3). Это взаимодействие оказывает сильное влияние на функциональные свойства белков, поскольку взаимодействие между РС-1 и РС-2 уменьшает способность РС-1 активировать белки G [19].
Механизм образования кист в почках
Почки у пациентов с АДПКП, которые наследуют одну мутированную копию РС-1 или РС-2 от родителя, будут развиваться и нормально функционировать всю взрослую жизнь. Однако со временем в почках этих пациентов будут развиваться кисты. В некоторых исследованиях подтверждается, что линии клеток теряют обе функциональные копии гена полицистина. Это показывает, что дополнительная, «вторая» соматическая мутация может быть причиной образования кист. Согласно этой модели, каждая киста возникает как следствие явного события соматической мутации, что объясняет медленное прогрессирование болезни в течение десятилетий [20]. На прогрессирование заболевания могут влиять такие факторы, как уровень экспрессии белка РС-1, патогенные аллели и стадии развития почек при мутации РС-1. Потеря функциональности белков РС-1 и РС-2 в развивающейся почке вызывает гораздо более серьезное кистозное заболевание, чем во взрослой [21]. Эти данные свидетельствуют о том, что потеря функции полицистина в течение периода быстрого клеточного роста и деления, которая характеризует постнатальное развитие почек, создает предрасположенность к цистогенезу, тогда как функция полицистина после этого периода клеточной пролиферации гораздо менее критична. Медленный рост кист в течение взрослой жизни может объясняться инактивацией вторичной мутации. Ввиду важности РС-1 и РС-2 для роста и дифференцировки клеток снижение уровня функциональных полицистиновых белков, присутствующих в клетках индивидуумов, гетерозиготных по мутации АДПКП, может способствовать нарушению восстановительного процесса и образованию кисты. Подтверждением этому является кистозное заболевание у мышей, подвергшихся повреждению почек, что привело к инициации воспаления и делению клеток. Следствием прогресссирования АДПКП является образование заполненных жидкостью кист, которые представляют собой резкий контраст с компактно расположенными канальцами в здоровой почке. На клеточном уровне эта трансформация приводит к следующим изменениям. Клетки организовываются в сферические, а не тубулярные структуры, и их полость заполняется жидкостью для расширения следующих кист (рис. 4). Кисты увеличиваются скорее за счет повышения количества клеток, чем за счет растягивания эпителиального слоя [22]. Одна из моделей, демонстрирующих переход от тубулярной формы к сферической, подтверждает, что нарушение полярности клеток приводит к тому, что тубулярные клетки больше не делятся вдоль оси параллельно канальцам.
/19-1.jpg)
Тщательный анализ прекистовых тубул на моделях мышей, у которых отмечается специфическая почечная инактивация PKD1 и PKD2, показал, что клетки, выстилающие кистозные канальцы, теряют ориентационное деление после того, как канальцы начинают расширяться [23]. Дезориентация клеточного деления недостаточна для развития кисты. У мышей с мутацией реснитчатого фиброцистинового белка изменялась митотическая ориентация, но не образовывались кисты почек, потому что клетки, которые делятся в эпителиальной плоскости, мигрируют обратно в тубулы. Вероятно, дефекты клеточной полярности играют роль в образовании кист, однако, теряя эту полярность, не могут привести к образованию кисты. Еще одним фактором образования кисты служит расширение объема ее содержимого. При этом происходит преобразование кистозной оболочки и становится возможной ионная секреция эпителием.
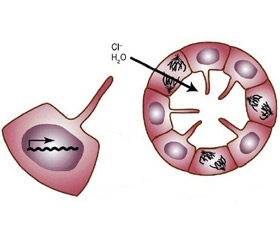
Секреция ионов вызывает парацеллюлярное или трансцеллюлярное осмотическое движение воды в кисту. Основным компонентом этой секреции является транспорт ионов хлора (Cl–), стимулированный циклическим аденозинмонофосфатом (цАМФ). Движение жидкости, приводящее к образованию кисты, осуществляется посредством цАМФ и включает трансмембранный регулятор апикального кистозного фиброза и базолатеральный Na-K-2Cl сопереносчик [24]. В эпителии Na-K-2Cl сопереносчик неактивен. Его активации при АДПКП на базальной мембране клеток способствует дополнительное поступление ионов натрия и хлора в кисту. РС-1 может влиять на экспрессию, локализацию или активность каналов Cl–. Экспрессия только С-концевого остатка усиливает транспорт Cl–, пролонгируя АТФ-стимулированную проводимость Cl–.
Необходимо отметить важную роль эпидермального фактора роста (EGF), который участвует в пролиферации клеток и экспансии почечных кист. Эпителиальные клетки кист чрезвычайно восприимчивы к пролиферативному стимулу EGF. Кроме того, кистозная жидкость содержит митогенные концентрации EGF, что может индуцировать клетки к пролиферации. Рецепторы EGF (EGFR) в норме локализованы на базолатеральной поверхности эпителиальных клеток собирательных канальцев почек. Однако у пациентов с АДПКП рецепторы EGF находятся на апикальной поверхности клеток собирательных канальцев. Эта неправильная локализация к апикальной поверхности сопровождается увеличением тирозинкиназной активности EGFR, что служит триггером для клеточной пролиферации и приводит к образованию и расширению кист в собирательных канальцах. Снижение EGFR тирозинкиназной активности приводит не только к уменьшению кисты, но и к значительному улучшению тубулярной функции путем концентрирования мочи. Сверхэкспрессия EGFR в собирательных канальцах может служить многообещающей мишенью для терапевтических лекарственных препаратов при лечении АДПКП.
Белки полицистина могут регулировать уровень цАМФ, так как кистозная болезнь связана с неправильным регулированием фосфодиэстеразы, которая разрушает цАМФ (рис. 5).
Роль первичных ресничек в патогенезе поликистозного заболевания почек
АДПКП является патологией, связанной с дисфункцией ресничек, известной также как цилиопатия [25, 26]. Первичная ресничка представляет собой одиночную «9 + 0» (не имеют двух синглетных микротрубочек в центре) микротрубочку, подобную волоску, прикрепленную к материнской центриоли и выступающую с поверхности почти всех клеток млекопитающих. Кроме широкого спектра сенсорных, первичные реснички выполняют ряд важных физиологических функций. Впервые они были описаны K. Zimmermann еще в 1898 г. [27]. С тех пор первичные реснички считались нефункциональными ремнантами, оставшимися от эволюции. Это представление о ресничках сохранялось до недавнего времени. Однако начиная с 2000 г. многочисленные исследования были сфокусированы на этой органелле, ее структуре и функционировании. В своей структуре первичная ресничка имеет 5 компартментов (рис. 6).
Аксонема состоит из 9 параллельных пар микротрубочек, которые посттрансляционно ацетилированы для поддержания длинной структуры. Эти микротрубочки расположены по окружности без центральной пары, которая присутствует в подвижных ресничках. Цилиарная мембрана содержит множество рецепторов, ионных каналов, транспортеров и сенсорных белков, выполняющих определенные функции. Некоторые из рецепторов локализуются на мембране реснички только в определенное время для выполнения соответствущей функции, а затем вытесняются из реснички. Цилиоплазма состоит из растворимого компартмента цилии и действует как кальциевый сигнализатор в ответ на механические или химические раздражители [28]. Кроме того, цилиоплазма обогащена многими сигнальными белками. Этот динамический компартмент включает в себя в основном два типа белков — сигнальные и транспортные. Данные белки необходимы для координации ключевой роли в построении и функционировании ресничек. Базальное тело — это материнская центриоль, с которой прочно соединена цилиарная аксонема. Помимо своей важной структурной роли базальное тело содержит много сигнальных белков, выполняющих различные функции. Область переходной зоны состоит непосредственно из самой переходной зоны и волокон. Эта область связывает базальное тело и цилиарную аксонему и играет важную роль в цилиогенезе и цилиарном доступе.
Первичные реснички, находящиеся на поверхности почечных эпителиальных клеток, являются механосенсорными органеллами, которые инициируют широкий спектр передачи Са2+-опосредованных регуляторных сигналов, т.е. реснички «отслеживают» скорость потока мочи через почечные канальцы. Комплекс белков РС-1 и РС-2, которые локализованы на поверхности первичной реснички, действует как кальциевый канал. Поток во внутрипочечных канальцах вызывает изгиб ресничек, который приводит к потоку кальция и интрацеллюлярному высвобождению кальция (рис. 7). В ответ на это происходит инициация триггерной молекулярной активности внутри клетки, которая регулирует развитие ткани и гомеостаз [29].
/20-1.jpg)
В результате воздействия на первичную ресничку механических стимулов происходит инициация входа Са2+ в цитоплазму, что запускает каскад сигналов, регулирующих пролиферацию клеток (IFT — intraflagellar transport — представляет собой главный транспортный механизм цилиарного компонента, необходимый для построения и стационарной поддержки ресничек). Ток жидкости по почечным канальцам вызывает наклон аксонемы, повышая концентрацию внутриклеточного Са2+, в то время как при нарушении функции ресничек этого не происходит.
В исследованиях S. Lee и соавт. [30] предложена новая гипотеза реснитчато-зависимого роста кисты при аутомосмно-доминантном поликистозе почек (рис. 8).
Таким образом, нарушение функционирования или полная потеря первичной реснички, сопровождающаяся нарушением восприимчивости почечного эпителия к току мочи и нарушением гомео–стаза Са2+, способствует расстройству регуляции клеточного цикла, что ведет к неконтролируемой пролиферации клеток и формированию кист.
Лечение аутосомно-доминантного поликистоза почек
Хотя поликистозное заболевание почек, проявлением которого могут быть гематурия, гипретония, образование камней в почках, аневризм сосудов головного мозга, дивертикулез толстой кишки, метаболические нарушения, пролапс митрального клапана, синдром поликистозных яичников, хроническая почечная недостаточность, известно уже более 300 лет, до настоящего времени специфического лечения этой патологии не существует. Благодаря современным достижениям в области медицины сейчас АДПКП диагностируется на ранних стадиях и более часто. Существует несколько стратегий, с помощью которых качество и продолжительность жизни пациентов улучшились. К ним относятся раннее выявление и лечение гипертонии, модификация образа жизни, лечение почечных и непочечных осложнений, хронической болезни почек и почечная заместительная терапия. Однако подходы к диагностике, лечению и профилактике АДПКП существенно различаются между странами и внутри страны. В настоящее время общепринятых практических рекомендаций по ведению пациентов с АДПКП не существует [31]. Основные исследования АДПКП экспотенциально увеличились за последние 30 лет, особенно после обнаружения генов PKD1 (1994 г.) и PKD2 (1996 г.), благодаря чему стала доступна молекулярно-генетическая диагностика. Несмотря на отсутствие одобренного FDA лечения этой болезни, был достигнут существенный прогресс в понимании клеточных сигнальных путей, ответственных за патогенез. Благодаря этому были определены новые потенциальные лекарственные средства для замедления или остановки прогрессиирования заболевания. Многие терапевтические препараты были испытаны на животных, что дало обнадеживающие результаты.
Наиболее перспективные лекарственные препараты для лечения АДПКП представлены в табл. 3.
1. Антагонисты рецептора вазопрессина
Вазопрессин, также известный как антидиуретический гормон, представляет собой нейрогипофизный гормон, состоящий из 9 аминокислот. У большинства видов он содержит аргинин и поэтому называется аргинин-вазопрессином. Гормон вазопрессин выполняет две основные функции — удержание воды в организме за счет увеличения ее реабсорбции в дистальных канальцах, а также собирательных трубочках нефронов почек и сужение кровеносных сосудов. Вазопрессин проявляет свои эффекты через рецепторы V1 и V2. Стимуляция V2-рецепторов базолатеральной мембраны клеток дистальных отделов почечных канальцев через цАМФ, являющийся вторичным мессенджером, вызывает повышение проницаемости стенки канальцев для воды, ее реабсорбцию и концентрирование мочи. Механизм действия гормона вазопрессина состоит в следующем. В крови на базолатеральной мембране клеток эпителия почечного канальца он связывается с V2-рецептором, что приводит к активации аденилатциклазы, образованию цАМФ и активации протеинкиназы А. Последняя вызывает фосфорилирование молекул аквапорина-2 в цитоплазматических пузырьках, их транспорт к апикальной мембране, где особые рецепторные молекулы обеспечивают встраивание молекул аквапорина-2 в мембрану и формирование водных каналов.
Было показано, что у мышей с поликистозным заболеванием почек происходит увеличение уровня циркулирующего вазопрессина. Многие кисты, полученные из собирающих протоков, экспрессируют рецепторы вазопрессина V2. Вазопрессин связывается с рецепторами V2 и активирует аденилатциклазу, что приводит к повышению уровня цАМФ, который способствует образованию кисты, стимулируя секрецию жидкости и пролиферативную активность кисты эпителиальных клеток. Доклинические исследования с ОРС-31260, антагонистом V2-рецептора, на моделях ортологичных мышей показали значительное ингибирование прогрессирования заболевания, что выражалось в уменьшении общего объема почек (ООП), кисты, митотических и апоптотических показателей, а также уровня азота мочевины [32]. Один из антагонистов рецептора V2 — ОРС-41061, или толваптан, c более высокой аффинностью, был испытан на ортологичных моделях животных с АДПКП и проявил высокую эффективность.
Лекарственный препарат толваптан (Samsca®) компании Otsuka Pharmaceutical Co., Ltd, Япония, является селективным ингибитором V2-рецепторов вазопрессина, замедляющим прогрессирование АДПКП, подавляет рост и развитие кист.
Механизм действия толваптана
Толваптан является антагонистом вазопрессина и имеет повышенную аффинность и селективность к V2-рецептору. Антагонизм к V2-рецептору проявляется уменьшением количества аквапорин-2-каналов в собирательных канальцах почек, что приводит к уменьшению реабсорбции воды, приросту выделения свободной воды (акварезис) и увеличению концентрации натрия в сыворотке [33] (рис. 9).
Исследование толвапатана было проведено в мультицентровом, двойном, слепом, плацебо-контролируемом параллельном исследовании TEMPO 3:4 (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes). В исследование включались пациенты из Северной и Южной Америки, Европы, Японии и Австралии. Первичной конечной точкой являлась оценка долгосрочной эффективности толваптана при АДПКП по изменению скорости ООП в процентах у пациентов, принимающих толваптан, по сравнению с пациентами, получающих плацебо (пациенты: 18–50 лет, общий объем почки ≥ 750 мл, скорость клубочковой фильтрации ≥ 60 мл). Вторичная конечная точка — оценка долгосрочной эффективности толвапатана с применением клинических маркеров прогрессирования АДПКП, таких как гипертензия, боль в почке, альбуминурия и почечная функция [34] (рис. 10).
При введении толваптана в течение 36 месяцев отмечались замедление роста ООП и функциональное снижение и уменьшение частоты осложнений, связанных с АДПКП. Изменение ООП при использовании толваптана по сравнению с плацебо было более выраженным в первый год лечения. В течение 3-летнего периода исследования TEMPO 3:4 прием толваптана приводил к уменьшению ООП с 5,5 % в год (плацебо) до 2,8 % в год [35] (рис. 11).
При анализе стадий хронической болезни почек (ХБП) было показано, что толваптан уменьшал скорость роста почек при различных стадиях ХБП (рис. 12).
Исследование TEMPO 3:4 продемонстрировало значительное влияние антагониста рецептора вазопрессина-2 толваптана на скорость роста ООП и скорость клубочковой фильтрации. На основе данных исследований толваптан был одобрен для применения в Японии и Канаде, а также рекомендован в Европейском союзе. В США FDA (Food and Drug Administration) запросило дополнительные данные для дальнейшей оценки эффективности и безопасности этого лекарственного препарата.
Таким образом, ключевое клиническое исследование TEMPO 3:4 для лечения АДПКП толваптаном показало следующую эффективность:
— замедление прогрессирования увеличения ООП;
— замедление ухудшения функции почек;
— улучшение клинических симптомов (боль).
В ходе основного исследования TEMPO 3:4 была обнаружена гепатотоксичность толваптана. В связи с этим FDA ввело ограничение на длительность использования данного препарата. Толваптан не должен применяться более 30 дней. Необходимо избегать применения препарата у пациентов с заболеваниями печени, включая цирроз.
2. Октреотид
Мутация при АДПКП приводит к снижению уровня внутриклеточного кальция и увеличению цAMФ, являющихся ключевыми внутриклеточными вторичными мессенджерами, которые вызывают пролиферацию клеток и секрецию жидкости в кисту. Один из этих вторичных мессенджеров — цАМФ — исследовали для лечения АДПКП. В качестве лечебного терапевтического препарата использовали аналог соматостатина октреотид.
Октреотид (Sandostatin®, Novartis Pharmaceuticals Corporation, Швейцария) представляет собой октапептид. Впервые был синтезирован в 1979 году химиком Вилфридом Бауэром.
Октреотид связывается с рецептором соматостатина и ингибирует продукцию цАМФ. Было показано, что октреотид снижает уровень цАМФ и ингибирует образование кист печени у крыс с поликистозным заболеванием почек, однако без какого-либо улучшения функции почек [36]. Небольшое рандомизированное плацебо-контролируемое исследование с участием 12 пациентов в течение 6 месяцев показало, что октреотид уменьшал ООП, но не улучшал СКФ [37]. Еще одним мультицентровым плацебо-контролируемым параллельным исследованием октреотида является испытание ALADIN, которое включало 79 пациентов с АДПКП. Целью данного исследования была оценка влияния действия октреотида в течение 3 лет на рост почек и кисты, а также оценка функций почек. Первичной конечной точкой являлось измерение ООП с помощью МРТ через один год и три года наблюдения [38]. Через год среднее изменение ООП составило 46,2 мл в группе октреотида и 143,7 мл в группе плацебо. Через 3 года среднее увеличение составило 220,1 мл в группе октреотида и 454,3 мл в группе плацебо. Существенной разницы в изменении СКФ от исходного уровня в течение 3 лет исследования не было.
/23-1.jpg)
3. Рапамицин
Рапамицин, или сиролимус, — иммунодепрессант, который применяется для борьбы с отторжением почки при трансплантации. Название «рапамицин» происходит от названия острова Пасхи (Рапа-Нуи). Рапамицин является продуктом бактерий вида Streptomyces hygroscopicus из образца почвы с этого острова. В ходе исследований выявились его иммуносупрессорные и антипролиферативные свойства.
Механизм действия рапамицина
Действие рапамицина направлено на mTOR (target of rapamycin), который является представителем семейства киназ, родственных фосфоинозитолкиназе, фосфорилирующей S6-киназу (S6K) и 4E-связывающий протеин (4EBP1). Фосфорилирование 4EBP1 высвобождает EIF4E, способствующий трансляции мРНК (рис. 14) Повышение активности mTOR и S6К обнаружено в клетках, выстилающих почечные кисты. Действие рапамицина замедляло прогрессирование наследственного поликистоза почек у крыс и мышей [39, 40]. Рапамицин индуцирует апоптоз эпителиальных клеток, окружающих кисты, и уменьшает их объем даже на поздних стадиях заболевания [41].
Цель 18-месячного открытого рандомизированного контролируемого исследования, опубликованного A. Serra и др. [42], заключалась в определении влияния сиролимуса на рост ООП у пациентов с АДПКП в возрасте от 18 до 40 лет, которые были рандомизированы для получения сиролимуса (целевая доза 2 мг/день) или стандартного лечения. Ключевые результаты данного исследования:
— не обнаружено различий между сиролимусом и стандартным лечением при исследовании ООП в течение 18 месяцев (первичная точка);
— нет значительных различий в СКФ.
Цель другого 2-летнего двойного слепого испытания состояла в том, чтобы исследовать влияние лечения эверолимусом на изменение ООП у пациентов с АДПКП [43]. В этом исследовании ООП через 12 месяцев увеличился на 102 мл в группе эверолимуса и 157 мл в группе плацебо. Однако через 2 года статистических значимых различий не было. Изменение от исходного уровня после 24 месяцев составляло 230 мл у получавших эверолимус и 301 мл в группе плацебо. Среднее значение расчетной скорости клубочковой фильтрации через 2 года составило 8,9 мл/мин/1,73 м2 в группе эверолимуса и 7,7 мл/мин/1,73 м2 в группе плацебо. Частота серьезных побочных эффектов составила 37,4 % при использовании эверолимуса и 23,5 % — плацебо. Соотношение пациентов, которые выбыли из группы исследования, составило 32,7 % в группе эверолимуса и 23,7 % в группе плацебо.
4. Ренин-ангиотензин-альдостероновая система
Известно, что ангиотензин II, помимо его вазоконстрикторного эффекта, увеличивает клеточную пролиферацию, ангиогенез, фиброз, известные как почечные компоненты при АДПКП. Потенциальные косвенные эффекты ангиотензина II на гипертонию включают стимуляцию симпатической нервной системы, эндотелия и альдостерона с удержанием натрия. Ангиотензин II также стимулирует реактивные формы кислорода, которые могут приводить к эндотелиальной дисфункции при АДПКП [45] (рис. 15).
Гипертензия присутствует при АДПКП и связана с увеличением ОПП, активацией РААС и прогрессированием заболевания почек.
В двойное слепое плацебо-контролируемое исследование [46] было включено 558 участников с АДПКП (от 15 до 49 лет, СКФ > 60 мл/мин/1,73 м2) со стандартным давлением крови от 120/70 до 130/80 мм рт.ст. или низким давлением крови от 96/60 до 110/75 мм рт.ст., которые получали ингибитор ангиотензинпревращающего фермента (лизиноприл) плюс блокатор рецептора ангиотензина (телмисартан) или лизиноприл плюс плацебо. Первичный результат оценивали по ежегодному процентному изменению ООП. Исходный уровень ООП составлял 1185 мл в группе пациентов с низким целевым давлением крови и 1240 мл в стандартной целевой группе. В течение 5 лет исследования наблюдали значительные различия в группе с низким целевым давлением. Рост ООП в этой группе составил 5,6 % в год, а в группе со стандартным давлением крови — 6,6 % в год. В относительном выражении это привело к 14,2% медленному ежегодному увеличению ООП для низкой целевой группы по сравнению со стандартной. За 5 лет исследования увеличение ООП в низкой целевой группе составило 38 % по сравнению с 44,25 % в стандартной. При этом не было существенной разницы между группами в отношении расчетной СКФ. Среднее ежегодное снижение СКФ составило 2,9 мл/мин/1,73 м2 у пациентов низкой целевой группы и 3,0 мл/мин/1,73 м2 — стандартной (рис. 16).
/24-1.jpg)
Одним из главных факторов риска прогрессирования повреждения почек при АДПКП считается гипертензия. Дефекты первичной реснички вызывают эндотелиальную дисфункцию и активацию РААС, являющиеся центральным патофизиологическим звеном развития гипертензии при АДПКП. Лечение гипретензии может замедлить прогрессирование болезни почек. Кроме диеты и модификации образа жизни первым антигипертензивным средством являются ингибиторы ангиотензинпревращающего фермента (иАПФ). Бета-блокаторы также эффективны для снижения артериального давления у пациентов с АДПКП. Их применение может быть особенно полезно у пацентов с АДПКП, у которых значительно повышен уровень концентрации креатинина в сыворотке. Бета-блокаторы могут быть лучшим выбором, чем блокаторы кальциевых каналов, так как они имеют более мягкие РААС-ингибиторные свойства. Диуретики следует рассматривать только в сочетании с ингибиторами РААС и мониторингом креатинина сыворотки, особенно у пациентов с выраженной почечной недостаточностью, когда риск острого ухудшения функции почек при комбинированной терапии с применением мочегонных средств становится выше. В настоящее время у пацентов с гипертонической болезнью АДПКП рекомендуется целевое давление крови < 130/80 мм рт.ст.
5. Статины
В экспериментальных моделях было показано, что статины могут ослабить прогрессирующую нефропатию [47]. Механизм действия ингибиторов HMG-СоА редуктазы (статины) на АДПКП в настоящее время неизвестен. Предполагается, что он не зависит от снижения уровня холестерина.
Рандомизированное двойное слепое плацебо-контролируемое клиническое исследование III фазы заключалось в оценке влияния правастатина на ООП, индекс массы левого желудочка и определении альбумина в моче у детей и молодых людей с АДПКП. Субъектами были 110 педиатрических пациентов с АДПКП и нормальной функцией почек, получающих лизиноприл. Они были рандомизированы в группу для лечения правастатином (20–40 мг/день) и в группу плацебо в течение 3 лет с оценкой через 18 и 36 месяцев [48]. Исходный уровень ООП составлял в группе правастатина 571 мл, в группе плацебо — 522 мл.
Ключевым итогом исследования стало то, что в группе правастатина увеличение ООП было значительно ниже по сравнению с плацебо (23 % против 33 %) (рис. 17).
6. Росковитин — ингибитор циклинзависимой киназы
Циклинзависимая киназа (CDK5) регулирует длину первичной реснички, влияя на динамику тубулина через его субстрат, известный как коллапсин [53]. Функция CDK5 при поликистозной болезни почек была установлена у мутантных молодых мышей jck с инактивированным геном CDK5. Первичные реснички эпителиальных клеток почек были увеличены у мышей jck по сравнению с контрольными мышами дикого типа. Нокдаун CDK5 привел к значительному сокращению длины реснички. Фармакологическое ингибирование CDK5 путем восстановления клеточной дифференцировки оценивали с использованием специфического ингибитора R-росковитина (проявляет высокую эффективность и селективность по отношению к некоторым циклинзависимым киназам). Этот ингибитор заметно уменьшал длину ресничек до уровней, сравнимых с контролем. Потеря CDK5 привела к сокращению длины ресничек и, соответственно, к уменьшению цистогенеза, улучшению функции почек и нормализации морфологии в кистозных эпителиальных клетках почек [54].
В эпителии кисты у мышей и человека с поликистозным заболеванием почек высока скорость пролиферации, что указывает на аномальное регулирование клеточного цикла. В исследовании N. Bukanov и др. [44] росковитин как ингибитор циклинзависимых киназ вводили двум моделям неортологических мышей jck и cpk. Росковитин приводил к остановке роста кисты у данных мышей. Более того, прерывистое введение этого препарата вызывало длительный антикистозный эффект. Учитывая хронический характер поликистозного заболевания почек, при котором может понадобиться пожизненная терапия, этот результат является многообещающим. Поэтому CDK5 является новой привлекательной терапевтической мишенью для лечения поликистозного заболевания почек.
7. Диета при АДПКП
Исследования на моделях животных с поликистозным заболеванием почек продемонстрировали, что на развитие этой патологии значительное влияние оказывает диета. Так, высокобелковая диета усугубляет цистогенез почек у мышей pcy и крыс. Диета с высоким содержанием белка была связана с низким аретриальным давлением, высоким уровнем внутриклеточного рН и неорганического фосфата, более высоким потреблением кислорода, образованием свбодных радикалов кислорода, снижением уровня глутатиона. Показано, что при изменении в диете белка изменяется активность почечной системы РААС и экспрессия трансформирующего фактора роста бета [49, 50]. Не только содержание белка, но и белковый состав диеты могут влиять на развитие поликистозного заболевания почек. Диета на основе соевого белка может благотворно влиять на развитие поликистозного заболевания почек у крыс и мышей по сравнению с диетой на основе казеина. Механизм, лежащий в основе этого важного эффекта, не был установлен. Соевый белок содержит несколько фитоэстрогенов или изофлавоноиды сои, включая генистеин, который является ингибитором тирозиновых протеинкиназ. Однако добавление генистеина в количестве, сравнимом с тем, которое присутствует в рационе на основе соевого белка, не оказало положительного эффекта. Поскольку соевый белок имеет высокое соотношение «аргинин/лизин» по сравнению с казеином, существует вероятность, что полезный эффект соевого белка связан с увеличением продуцирования оксида азота. Интересным является исследование, касающееся полезного действия соевого белка и его влияния на метаболизм полиненасыщенных жирных кислот [51]. Обнаружено, что благоприятный эффект соевого белка обусловлен уменьшением продуцирования арахидоновой кислоты. Введение соевого белка приводит к более низким уровням инсулиноподобного фактора роста-1 в почках. В отличие от исследований с использованием соевого белка и добавок льняного масла введение рыбьего жира, богатого источника омега-3-полиненасыщенных жирных кислот, оказывает долгосрочное влияние на функцию почек и выживаемость у мышей [52].
Выводы
1. Накоплен большой информационный материал о механизме АДПКП, определена структура белков PC-1/РС-2, их локализация и непосредственное участие в патогенезе данного заболевания.
2. АДПКП является патологией, связанной с дисфункцией ресничек.
3. Наиболее перспективными лекарственными препаратами для лечения АДПКП на сегодняшний день являются антагонисты рецептора вазопрессина-2, ингибиторы mTOR и цАМФ.
4. В настоящее время нет одобренного FDA фармакологического лечения данного заболевания и реальных перспектив патогенетической терапии АДПКП.
5. Лечение АДПКП рекомендуется начинать на ранней стадии заболевания, когда кисты малы, а функция почек хорошо сохраняется.
Конфликт интересов. Автор заявляет об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Spithoven E, Kramer A, Meijer E, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival-an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant. 2014 Sep;29 Suppl 4:iv15-25. doi: 10.1093/ndt/gfu017.
2. Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001 Oct 27;358(9291):1439-44. doi: 10.1016/S0140-6736(01)06531-X.
3. Wilson PD. Polycystic kidney disease. N Engl J Med. 2004 Jan 8;350(2):151-64. doi:10.1056/NEJMra022161.
4. Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995 Jun;10(2):151-60. doi:10.1038/ng0695-151.
5. Peters DJ, van de Wal A, Spruit L, et al. Cellular localization and tissue distribution of polycystin-1. J Pathol. 1999 Aug;188(4):439-46. doi:10.1002/(SICI)1096-9896(199908)188:4<439::AID-PATH367>3.0.CO;2-P.
6. Chauvet V, Qian F, Boute N, Cai Y, et. al. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am J Pathol. 2002 Mar;160(3):973-83. doi:10.1016/S0002-9440(10)64919-X.
7. Qian F, Boletta A, Bhunia AK, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A. 2002 Dec 24;99(26):16981-6. PMID: 12482949.
8. Xu C, Rossetti S, Jiang L, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007 Mar;292(3):F930-45. doi:10.1152/ajprenal.00285.2006.
9. Low SH, Vasanth S, Larson CH, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease Dev Cell. 2006 Jan;10(1):57-69. doi:10.1016/j.devcel.2005.12.005.
10. Köttgen M, Walz G. Subcellular localization and trafficking of polycystins. Pflugers Arch. 2005 Oct;451(1):286-93. doi:10.1007/s00424-005-1417-3.
11. Pelucchi B, Aguiari G, Pignatelli A, et al. Nonspecific cation current associated with native polycystin-2 in HEK-293 cells. J Am Soc Nephrol. 2006 Feb;17(2):388-97. doi:10.1681/ASN.2004121146.
12. Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003 Feb;33(2):129-37. doi:10.1038/ng1076.
13. Mochizuki T, Wu G, Hayashi T, at al. 1996. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996 May 31;272(5266):1339-42. doi:10.1126/science.272.5266.1339.
14. Koulen P1, Cai Y, Geng L, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002 Mar;4(3):191-7. doi:10.1038/ncb754.
15. Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007 Apr 10;104(15):6454-9. doi:10.1073/ pnas.0610324104.
16. Yoder BK1, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are colocalized in renal cilia. J Am Soc Nephrol. 2002 Oct;13(10):2508-16. doi:10.1097/01 .ASN.0000029587.47950.25.
17. Xu C, Rossetti S, Jiang L, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flowinduced Ca2+ signaling. Am J Physiol Renal Physiol. 2007 Mar;292(3):F930-45. doi:10.1152/ajprenal.00285.2006.
18. Casuscelli J, Schmidt S, DeGray B, et al. Analysis of the cytoplasmic interaction between polycystin-1 and polycystin-2. Am J Physiol Renal Physiol. 2009 Nov;297(5):F1310-5. doi: 10.1152/ajprenal.00412.2009.
19. Delmas P, Nomura H, Li X, Lakkis M, et al. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem. 2002 Mar 29;277(13):11276-83. doi:10.1074/jbc .M110483200.
20. Pritchard L, Sloane-Stanley JA, Sharpe JA, et al. A human PKD1 transgene generates functional polycystin-1 in mice and is associated with a cystic phenotype. Hum Mol Genet. 2000 Nov 1;9(18):2617-27. PMID: 11063721.
21. Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet. 2007 Dec 15;16(24):3188-96. doi:10.1093/hmg/ddm299.
22. Grantham JJ. The etiology, pathogenesis, and treatment of autosomal dominant polycystic kidney disease: recent advances. Am J Kidney Dis. 1996 Dec;28(6):788-803. doi:10.1016/S0272-6386(96)90378-9.
23. Nishio S, Tian X, Gallagher AR, et al. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol. 2010 Feb;21(2):295-302. doi: 10.1681/ASN.2009060603.
24. Montesano R, Ghzili H, Carrozzino F, Rossier BC, Féraille E. cAMP-dependent chloride secretion mediates tubule enlargement and cyst formation by cultured mammalian collecting duct cells. Am J Physiol Renal Physiol. 2009 Feb;296(2):F446-57. doi: 10.1152/ajprenal.90415.2008.
25. Abdul-Majeed S, Nauli SM. Polycystic diseases in visceral organs. Obstet Gynecol Int. 2011; 2011:609370. doi:10.1155/2011/609370.
26. Ratnam S, Nauli SM. Hypertension in Autosomal Dominant Polycystic Kidney Disease: A Clinical and Basic Science Perspective. Int J Nephrol Urol. 2010;2:294-308. PMCID: PMC4215423.
27. Zimmermann KW. Beiträge zur Kenntnis einiger Drüsen und Epithelien. Archiv f mikrosk Anat. 1898;52:552. doi: 10.1007/BF02975837. (in Germany).
28. Jin X, Mohieldin AM, Muntean BS, et al. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol Life Sci. 2014 Jun;71(11):2165-78. doi: 10.1007/s00018-013-1483-1.
29. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 2007;8(11):880-93. DOI:10.1038/nrm2278.
30. Lee SH, Somlo S. Cyst growth, polycystins, and primary ciliain autosomal dominant polycystic kidney disease. Kidney Res Clin Pract. 2014 Jun;33(2):73-8. doi: 10.1016/j.krcp.2014.05.002.
31. Chapman AB, Devuyst O, Eckardt K, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. 2015 International Society of Nephrology. Kidney Int. 2015 Jul;88(1):17-27. doi: 10.1038/ki.2015.59.
32. Wang X, Gattone V 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005 Apr;16(4):846-51. doi: 10.1681/ASN.2004121090.
33. Samsca (tolvaptan), package insert. Rockville, MD: Otsuka America Pharmaceutical, Inc, May 2009.
34. Torres VE, Meijer E, Bae KT, et al. Rationale and Design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes) 3-4 Study. Am J Kidney Dis. 2011 May;57(5):692-9. doi: 10.1053/j.ajkd.2010.11.029.
35. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012 Dec 20;367(25):2407-18. doi: 10.1056/NEJMoa1205511.
36. Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′, 5-cyclic monophosphate. Gastroenterology. 2007 Mar;132(3):1104-16. doi:10.1053/j.gastro.2006.12.039.
37. Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206-16. doi:10.1111/j.1523-1755.2005.00395.x.
38. Caroli A, Perico N, Perna A, et al. Effect of long acting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet 2013; 382(9903):1485-95. doi:10.1016/S0140-6736(13)61407-5.
39. Tao Y, Kim J, Schrier R, Edelstein CL. Rapamycin markedly slows disease progression in rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16(1):46-51. doi: 10.1681/ASN.2004080660.
40. Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wüthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2006 Mar;21(3):598-604. doi:10.1093/ndt/gfi181.
41. Shillingford J, Murcia N, Larson C, et al. The mTOR pathway is regulated by polycystin-1 and its inhibition reverses cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006 Apr 4;103(14):5466-71. doi:10.1073/pnas.0509694103.
42. Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):820-9. doi:10.1056/NEJMoa0907419.
43. Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9):830-40. doi:10.1056/NEJMoa1003491.
44. Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006 Dec 14;444(7121):949-52. doi:10.1038/nature05348.
45. Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009 Sep;20(9):1888-93. doi: 10.1681/ASN.2008080882.
46. Schrier RW, Abebe KZ, Perrone RD, et al. Blood Pressure in Early Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2014 Dec 11;371(24):2255-66. doi: 10.1056/NEJMoa1402685.
47. Hafez KS, Inman SR, Stowe NT, Novick AC. Renal hemodynamic effects of lovastatin in a renal ablation model. Urology 1996;48(6):862-7. PMID: 8973668.
48. Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9(5):889-96. doi:10.2215/CJN.08350813.
49. Tomobe K, Philbrick D, Aukema H, et al. Early dietary protein restriction slows disease progression and lengthens survival in mice with polycystic kidney disease J Am Soc Nephrol. 1994 Dec;5(6):1355-60. PMID: 7894002.
50. Ogborn M, Sareen S. Amelioration of polycystic kidney disease by modification of dietary protein intake in the rat. J Am Soc Nephrol. 1995 Dec;6(6):1649-54. PMID: 8749693.a
51. Ogborn M, Nitschmann E, Weiler H, Bankovic-Calic N. Modification of polycystic kidney disease and fatty acid status by soy protein diet. Kidney Int. 2000 Jan;57(1):159-66. doi: 10.1046/j.1523-1755.2000.00835.x.
52. Aukema H, Yamaguchi T, Takahashi H, et al. Effects of dietary fish oil on survival and renal fatty acid composition in murine polycystic kidney disease. Nutr Res. 1992;12:1383-92.
53. Ou Y, Zhang Y, Cheng M, Rattner JB, Dobrinski I, van der Hoorn FA. Targeting of CRMP-2 to the primary cilium is modulated by GSK-3ß. PLoS One. 2012;7(11):e48773. doi: 10.1371/journal.pone.0048773.
54. Husson H, Moreno S, Smith LA, et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophtisis. Hum Mol Genet. 2016 Jun 1;25(11):2245-55. doi:10.1093/hmg/ddw093.

/14-1.jpg)
/17-1.jpg)
/17-2.jpg)
/18-1.jpg)
/19-1.jpg)
/19-2.jpg)
/20-1.jpg)
/21-1.jpg)
/22-1.jpg)
/23-1.jpg)
/23-2.jpg)
/24-1.jpg)
/25-1.jpg)