Резюме
Мета. Аналіз особливостей клінічної картини хвороби Помпе (ХП) у хворих дитячого віку. Визначення за результатами лабораторних та інструментальних досліджень основних клініко-лабораторних характеристик, що дозволяють виділити таких пацієнтів у групу селективного скринінгу для проведення ферментної діагностики на наявність ХП у сухій краплі крові. Матеріали та методи. В період 2002–2016 рр. у Центрі орфанних захворювань НДСЛ «Охматдит» обстежені 522 дитини з підозрою на ХП. Вік пацієнтів — від 4 місяців до 13 років; хлопчиків було 282, дівчаток — 240. Результати. У 98 (19 %) хворих за даними електронейроміографії і на підставі виявлення мутацій у гені SMN діагностована спінальна м’язова атрофія, у 70 (13 %) — первинна м’язова дистрофія Дюшена. У 71 (13,6 %) пацієнта виявлені різні форми спадкової нейромотосенсорної невропатії. У 38 (7,2 %) пацієнтів відзначали домінантний тип успадкування захворювання, в подальшому лабораторні дослідження з метою підтвердження діагнозу ХП у них не проводили. Отже, у селективну (скринінгову) групу для проведення ферментодіагностики у сухій краплі крові для виявлення ХП включено 245 (47 %) осіб. У цих пацієнтів відзначали гетерогенну клінічну картину захворювання: м’язову слабкість, зумовлену міопатичним синдромом, у поєднанні з кардіоміопатією, затримкою фізичного розвитку, гіперкреатинфосфатемією. У 25 хворих віком 2–8 місяців, крім міопатичного синдрому, спостерігали підвищення рівня аланінамінотрансферази та аспартатамінотрансферази, креатинфосфокінази (понад 220 од/л), лактатдегідрогенази (понад 250 од/л), гіпертрофічну кардіоміопатію лівого шлуночка; у 10 з цих хворих виявлені гепатомегалія та серцево-легенева недостатність. За результатами ферментодіагностики, діагноз ХП (інфантильна форма) встановлений у 5 пацієнтів (у 3 — класична форма, у 2 — атипова, або некласична). Клінічна картина була гетерогенною, проте в усіх спостереженнях відзначали сповільнене збільшення маси тіла, затримку формування статокінетичних навичок. Три пацієнти отримували ферментозамісну терапію, що дало можливість запобігти ранній смерті у цих пацієнтів і в одному випадку практично відновити самостійну ходьбу. Висновки. ХП у дітей характеризується мультисистемним ураженням, різними клінічними формами, за відсутності специфічного лікування призводить до ранньої смерті хворих. Основними ознаками ХП у дітей є гіпертрофічна кардіоміопатія, міопатичний синдром у поєднанні зі специфічним біохімічним фенотипом. Ферментодіагностика у сухій краплі крові в групі селективного скринінгу може підвищити ефективність ранньої діагностики та лікування дітей при ХП.
Цель. Анализ особенностей клинической картины болезни Помпе (БП) у больных детского возраста. Определение по результатам лабораторных и инструментальных исследований основных клинико-лабораторных характеристик, позволяющих выделить таких пациентов в группу селективного скрининга для проведения ферментной диагностики на наличие БП в сухих каплях крови. Материалы и методы. В период с 2002 по 2016 г. в Центре орфанных заболеваний НДСБ «Охматдет» обследованы 522 ребенка с подозрением на БП. Возраст больных — от 4 месяцев до 13 лет, мальчиков было 282, девочек — 240. Результаты. У 98 (19 %) больных по данным электронейромиографии и на основания выявления мутаций в гене SMN диагностирована спинальная мышечная атрофия, у 70 (13 %) — первичная мышечная дистрофия Дюшена. У 71 (13,6 %) пациента выявлены различные формы наследственной нейросенсомоторной нейропатии. У 38 (7,2 %) пациентов наблюдался доминантный тип наследования заболевания, в дальнейшем лабораторные исследования с целью подтверждения диагноза БП у них не проводили. Таким образом, в селективную (скрининговую) группу для проведения ферментодиагностики в сухих каплях крови включены 245 (47 %) пациентов. У этих больных отмечена гетерогенная клиническая картина заболевания: мышечная слабость, обусловленная миопатическим синдромом в сочетании с кардиомиопатией, задержкой физического развития, гиперкреатинфосфатемией. У 25 больных в возрасте 2–18 месяцев, кроме миопатического синдрома, наблюдали повышение уровней аланинаминотрансферазы и аспартатаминотрансферазы, креатинфосфокиназы (свыше 220 ед/л), лактатдегидрогеназы (свыше 250 ед/л), гипертрофическую кардиомиопатию левого желудочка, у 10 из этих больных выявлены гепатомегалия и сердечно-легочная недостаточность. По результатам ферментодиагностики диагноз БП (инфантильная форма) установлен у 5 пациентов (у 3 — классическая форма, у 2 — атипичная, или неклассическая). Клиническая картина была гетерогенной, однако во всех наблюдениях отмечали замедленную прибавку массы тела, задержку формирования статокинетических навыков. Три пациента получали ферментозаместительную терапию, что позволило предотвратить раннюю смерть и в одном случае практически восстановить самостоятельную ходьбу. Выводы. БП у детей характеризуется мультисистемным поражением и различными клиническими формами, при отсутствии специфического лечения приводит к ранней смерти больных. Основными проявлениями БП у детей стали гипертрофическая кардиомиопатия, миопатический синдром в сочетании со специфическим биохимическим фенотипом. Ферментодиагностика в сухих каплях крови в группе селективного скрининга позволит повысить эффективность ранней диагностики и лечения детей при БП.
Background. Analysis of Pompe disease (PD) clinical features in children in order to determine its main clinical and laboratory characteristics based on the results of laboratory and instrumental studies that allow to select such patients into the screening group for enzymatic diagnosis in dried blood spots was the objective of this study. Materials and methods. In a period from 2002 to 2016, 522 children with PD suspicion were examined in the Center of Orphan Diseases of National Children’s Specialized Hospital “Ohmatdyt”. There were 282 boys and 240 girls aged from 4 months to 13 years. Results. According to electroneuromyography results and mutations in SMN gene revealing, spinal muscular atrophy was diagnosed in 98 (19 %) patients, Duchenne primary muscular dystrophy — in 70 (13 %). 71 (13.6 %) patients had different forms of hereditary neuropathy. In 38 (7.2 %) cases, the dominant type of disease inheritance was revealed, later we did not provide laboratory tests in those patients in order to confirm the PD diagnosis. Thus, 245 (47 %) patients were included into the selective (screening) group for enzymatic diagnosis using dried blood spot. In these patients, a heterogeneous clinical picture of the disease was noted: muscle weakness due to myopathic syndrome in combination with cardiomyopathy, delay in physical development, hyperCKemia. Twenty five patients aged 2 to 8 months besides myopathic syndrome had elevated levels of alanine aminotransferase and aspartate aminotransferase, creatine phosphokinase (more than 220 U/l), lactate dehydrogenase (more than 250 U/l), left ventricular hypertrophic cardiomyopathy; also in 10 patients, hepatomegaly and cardiopulmonary failure were revealed. Based on enzymatic tests, PD diagnosis (infantile form) was established in 5 patients (in 3 cases — classical form, in 2 cases — atypical). The clinical picture was heterogeneous, however, in all cases, slow increase in body weight, delay of static and kinetic skills forming were noted. Three patients receive enzyme replacement therapy. This allowed to prevent early death, and in one case, it was almost possible to restore independent walking. Conclusions. PD in children is characterized by different system failure and various clinical forms, in specific treatment absence, PD leads to patients’ early death. The PD main manifestations in children are hypertrophic cardiomyopathy, myopathic syndrome in combination with a specific biochemical phenotype. Enzymatic diagnosis in dried blood spots in the group of selective screening will increase the effectiveness of PD early diagnosis and treatment in children.
Робота проведена в рамках НДР «Визначення генетичних основ ризику розвитку патологічних станів на різних етапах онтогенезу», номер держреєстрації 0114U002215 (2014–2018 рр.).
Вступ
Хвороба Помпе (ХП) (OMIM232300) — рідкісне (орфанне) спадкове захворювання, яке належить до лізосомальних хвороб накопичення. За характером субстрату, що накопичується, ХП зараховують до глікогенозів II типу, за клінічними проявами — до прогресуючих нервово-м’язових захворювань. В основі патогенезу ХП лежить зниження активності (або цілковита її відсутність) ферменту лізосом — кислої α-глюкозидази (GAA), що бере участь у розщепленні глікогену. Через низьку активність ферменту GAA нерозщеплений глікоген накопичується у лізосомах різних клітин, переважно у скелетних м’язах і міокарді, що призводить до механічної руйнації та загибелі міоцитів [1].
За даними американських і європейських дослідників, ХП становить близько 15 % у структурі усіх хвороб накопичення глікогену, яких на сьогодні виділяють 14 типів [2].
Механізм успадкування ХП — автосомно-рецесивний. Ген, який кодує фермент GAA, картований на хромосомі 17q25.2-q25.3., складається з 20 екзонів. Описані 360 мутацій, які спричиняють часткове або цілковите блокування каталітичної активності ферменту, зниження його стабільності та скорочення періоду напіврозпаду [3].
ХП виявляють в 1 випадку на 40 000 живих новонароджених, проте, за останніми даними, частота виявлення захворювання може варіювати в різних етнічних популяціях від 1 : 14 000 до 1 : 300 000 живих новонароджених [4].
ХП характеризується численними симптомами та варіантами перебігу. Із 1932 р. і дотепер чітко клінічно описана лише інфантильна форма ХП (ІФХП) [5].
Широко використовують класифікацію, в основу якої покладений вік хворого, в якому виникли перші симптоми захворювання: ІФХП або ХП із раннім (ХПРП) і пізнім (ХППП) початком [6]. ХПРП (ІФХП) має два клінічних варіанти: класичний і атиповий (некласичний). Класична ІФХП клінічно проявляється прогресуючою міопатією з генералізованою м’язовою слабкістю, гіпотонією, гіпертрофічною кардіоміопатією. За відсутності лікування у хворих виникає серцево-легенева недостатність; тривалість життя таких пацієнтів близько 8 місяців.
Некласична ІФХП характеризується незначними порушеннями з боку серця (інколи вони взагалі відсутні), певною затримкою формування моторних функцій на тлі гіпотонії та генералізованої м’язової слабкості. За відсутності лікування такі хворі живуть близько 2 років [7].
При ХППП перші ознаки захворювання виявляють у хворих віком 20–60 років. Пацієнти страждають від прогресуючої м’язової слабкості, зазвичай без суттєвих змін з боку серця. У міру прогресування захворювання поступово виникає дихальна недостатність, пацієнти потребують штучної вентиляції легень. Саме дихальна недостатність спричиняє ранню смерть таких хворих [8].
Золотим стандартом лабораторної діагностики при ХП вважають визначення активності ферменту GAA у лейкоцитах або культурі фібробластів шкіри пацієнта [10]. Рівень активності ферменту GAA у фібробластах шкіри пацієнтів при ХППП становить 1–40 % норми [9].
Крім того, останніми роками розроблений і широко застосовується метод визначення активності GAA у сухій краплі крові (СКК), який можна використовувати для селективного та масового неонатального скринінгу [11]. Результати молекулярного аналізу гена GAA важливі для медико-генетичного консультування та планування вагітності в обтяжених родинах.
У 2006 р. Food and Drug Administration (FDA) і European Medicines Agency (EMA) зареєстрували препарат алглюкозидаза альфа для лікування ХП [12]. Враховуючи наявність ефективних методів лікування ХП, надзвичайно важливим є питання ранньої діагностики захворювання.
Мета дослідження: проаналізувати дані клінічних, інструментальних і лабораторних особливостей клінічної картини у хворих на ХП; запропонувати алгоритм для оптимізації діагностики цього рідкісного захворювання у дітей.
Матеріали та методи
У Центрі орфанних захворювань НДСЛ «Охматдит» (2002–2016 рр.) обстежені 522 дитини з підо–зрою на ХП віком від 4 місяців до 13 років, хлопчиків було 282, дівчаток — 240.
Оцінювали загальний соматичний і неврологічний статус хворих, проводили електрокардіографію (ЕКГ), ехокардіографію (ЕхоКГ), елекронейроміо–графію (ЕНМГ), рентгенографію органів грудної клітки, біохімічні дослідження, зокрема визначення рівня креатинфосфокінази (КФК), печінкових трансаміназ — аланінамінотрансферази (АЛТ) і аспартатамінотрансферази (АСТ). Беручи до уваги наявність у клінічній картині міопатичного синдрому, для верифікації його форми у дітей молодше 5 років обов’язковим було проведення ДНК-аналізу з метою виключення спінальної м’язової атрофії та міопатії Дюшена. Молекулярно-генетичне дослідження проводили у приватних лабораторіях.
Основними критеріями відбору пацієнтів для проведення ферментодіагностики методом СКК були: наявність гіпертрофічної кардіоміопатії з серцево-легеневою недостатністю (або без такої), затримка фізичного та статомоторного розвитку, міопатичний синдром у дітей віком менше 5 років, підвищення рівня КФК і печінкових трансаміназ, а також результати клініко-генеалогічного аналізу, оскільки ХП успадковується винятково за автосомно-рецесивним типом. Такий підхід дозволив виключити з групи скринінгу пацієнтів із різноманітними нервово-м’язовими захворюваннями, що успадковуються за автосомно-домінантним типом.
У всіх хворих проведені комплексне клініко-лабораторне та інструментальне дослідження, генеалогічний аналіз. Визначали рівень КФК, АЛТ, АСТ, лактатдегідрогенази (ЛДГ). У 464 (75,6 %) пацієнтів виконана ЕНМГ, у 350 (57 %) — ЕКГ і ЕхоКГ. За наявності показань (згідно з результатами клініко-лабораторного дослідження) проводили молекулярно-генетичний аналіз на мутації в генах SMN і дистрофіну (Інститут молекулярної біології та генетики НАН України), молекулярно-генетичний аналіз на мутації в гені GAA (лабораторія Archimed Life Science GmbH, Відень, Австрія); визначали активність ферменту GAA у СКК (медико-генетична лабораторія НДСЛ «Охматдит», Україна) за модифікованим протоколом N.A. Chamоles і співавт. [13, 14] з використанням плашкового аналізатора Wallac Vіctоr TM 1420 Multіlabel Cоunter (Perkіn Elmer, Турку, Фінляндія).
Результати та обговорення
У 98 (19 %) хворих за даними ЕНМГ і на підставі виявлених у гені SMN мутацій діагностована спінальна м’язова атрофія, у 70 (13 %) — первинна м’язова дистрофія Дюшена. У 71 (13,6 %) хворого відзначені різні форми спадкової нейромотосенсорної невропатії. У 38 (7,2 %) пацієнтів відзначали домінантний тип успадкування захворювання, в подальшому лабораторні дослідження з метою підтвердження діагнозу ХП у них не проводили. Таким чином, у селективну (скринінгову) групу для ферментодіагностики у СКК з метою виявлення ХП включено 245 (47 %) хворих із гетерогенною клінічною картиною захворювання: м’язовою слабкістю, зумовленою міопатичним синдромом, у поєднанні з кардіоміопатією, затримкою фізичного розвитку, гіперкреатинфосфокімією. У 25 хворих віком 2–8 місяців, крім міопатичного синдрому, відзначали підвищення рівня АЛТ і АСТ, КФК (понад 220 од/л), ЛДГ (понад 250 од/л), гіпертрофію міокарда лівого шлуночка, у 10 з цих хворих виявлено гепатомегалію та серцево-легеневу недостатність.
За результатами ферментодіагностики діагноз ІФХП встановлений у 5 пацієнтів (у 3 — класична форма, у 2 — атипова або некласична). Клінічна картина захворювання була гетерогенною, проте в усіх хворих відзначали сповільнення збільшення маси тіла, затримку формування статокінетичних навичок (табл. 1).
Усі пацієнти народилися від нормальних вагітностей і фізіологічних пологів, антропометричні показники при народженні — у межах популяційної норми.
В усіх спостереженнях діагноз встановлений із затримкою: за класичного варіанта ІФХП — вік хворих у середньому становив 5,1 міс., за некласичного — 6,5 року, що характерно для цієї патології і знайшло відображення у працях інших дослідників [15].
В усіх пацієнтів відзначали порушення з боку шлунково-кишкового тракту — діарею, сповільнене збільшення маси тіла або її зменшення. У старших дітей спостерігали схильність до частих ГРВІ, що ускладнювалися виникненням дихальної недостатності та пневмонії. У 2 хворих через гострі порушення дихання встановлено трахеостому, ще 1 пацієнт потребував стаціонарної апаратної підтримки під час нічного сну. За даними літератури, прогресування ХП спричиняє фіброз легень, що зумовлює необхідність використання додаткових інструментальних методів моніторингу функції дихання (пульсоксиметрія, полісомнографія тощо) [16].
Одним з основних у клінічній картині захворювання був міопатичний синдром, вираженість якого різнилася залежно від форми ХП і віку пацієнтів. У хворих віком до 1 року спостерігали виражений міо–патичний синдром — генералізовану м’язову слабкість («млява» дитина), під час тракційного тесту — характерне закидання голови, симптом «провислої білизни». Для пацієнтів за класичної ХП характерні макроглосія та гіпомімія (табл. 2, рис. 1).
/72-2.jpg )
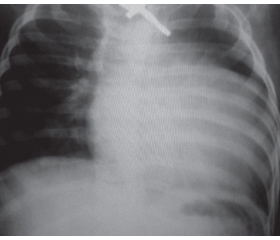
У дітей старшого віку за некласичного варіанта ІФХП на тлі гіпотрофії м’язового корсета відзначали виражену слабкість переважно у проксимальних групах м’язів кінцівок і тулуба, сколіоз, деформацію грудної клітки («серцевий горб»). За даними існуючих у світі реєстрів хворих на ХП, скелетні деформації виявляють майже у 50 % спостережень [15, 16]. Для класичного варіанта ІФХП характерний більш тяжкий і швидкий перебіг через порушення з боку серцево-судинної та дихальної систем. За даними рентгенографії грудної клітки, в усіх хворих за класичної ІФХП виявлене розширення меж серця з відповідними змінами на ЕКГ (високий вольтаж комплексу QRS, короткий інтервал PR) (рис. 2).
/73-1.jpg )
На момент встановлення діагнозу в усіх хворих при ІФХП відзначали поєднання вираженого міопатичного синдрому з прогресуючою гіпертрофічною кардіоміопатією, що спричиняла загрозливе зменшення фракції викиду крові з лівого шлуночка (від 67 до 45 %). У літературі описані спостереження виявлення ознак гіпертрофії лівого шлуночка серця у пренатальному періоді [17].
Отже, поєднання прогресуючої гіпертрофічної кардіоміопатії та міопатичного синдрому можна вважати діагностично значущим за класичної або інфантильної форми ХП.
Атиповий або некласичний варіант ХП виявлений у двох хворих віком 6 і 7 років. В обох спостереженнях ГРВІ спричинила виникнення спочатку гострих, а потім — хронічних розладів дихання. Обидва пацієнти потребували встановлення трахео–стоми та реанімаційної підтримки. Одну пацієнтку спостерігали з діагнозом «недиференційована форма міопатії», перші ознаки якої виникли у віці 6 місяців. ГРВІ у віці 6 років, майже через 5 років від дебюту захворювання, лише суттєво погіршила стан дитини: вона практично втратила здатність самостійно ходити, потребувала штучної вентиляції легень. В іншому спостереженні ознаки міопатичного синдрому були практично відсутні до виникнення ГРВІ. Проте виникнення гострих розладів дихання, а в подальшому — швидке прогресування слабкості м’язів тулуба і проксимальних відділів кінцівок, формування специфічної міопатичної ходи після гострого періоду ГРВІ стали справжньою несподіванкою. Специфічні кардіальні симптоми, притаманні класичній формі ХП, у цих двох хворих відсутні: в однієї дитини виникла кардіоміопатія легкого ступеня, у другої — несподівана знахідка — рабдоміома, що не притаманна ХП. Прояви кардіоміопатії суттєво зменшилися за два роки безперервної ферментозамісної терапії (ФТЗ).
За результатами біохімічних досліджень у всіх п’ятьох хворих відзначали стабільно високі рівні КФК, ЛДГ, АЛТ, АСТ. КФК — неспецифічний маркер міопатичних процесів різної етіології: запальних, спадкових (зокрема, дистрофінопатій тощо) і метаболічних (ХП, синдроми порушення мітохондріального бета-окислення та ін.). Рівень КФК при ХП не перевищує 2000 од/л, проте у деяких хворих (особливо при ХППП, ознаки якої виникли у дорослому віці) може бути в межах норми [18]. У нашому дослідженні рівень КФК не перевищував 1500 од/л. Кореляція між рівнем КФК, формою захворювання та ступенем його тяжкості відсутня [19].
У всіх хворих відзначали зниження остаточної активності GAА, що не перевищувала 0,23 нмоль/год/мг білка. Рівень активності GAA корелює зі ступенем тяжкості захворювання. Так, за цілковитої відсутності активності GAA перебіг ХП був найтяжчим, дитина померла у віці 4 місяців. Стан хворого за найвищого рівня активності GAA був тяжким, однак відзначали найбільш пізній дебют захворювання, збереження щоденної рухової активності, здатності самостійно ходити. Хворому встановлена трахеостома, проте він не потребував нічної апаратної підтримки дихання. Кореляцію між рівнем остаточної активності GAA, формою і ступенем тяжкості ХП пояснюють генно-фенотиповими особливостями. Отримані нами результати відповідають таким інших дослідників [17]. Хоча деякі автори не відзначали прямої кореляції між рівнем остаточної активності GAA і тяжкістю ХП [16].
При ХП час встановлення діагнозу є надзвичайно важливим, що пов’язано з ефективністю ФЗТ, яка запобігає необоротному ушкодженню м’язів, виникненню хронічних серцевих і легеневих порушень [2].
Молекулярно-генетичні дослідження проведені у 4 із 5 дітей. Спектр виявлених мутацій виявився унікальним. У пацієнта 2 виявлені нові мутації, не описані у базі даних мутацій в гені GAA, проте з огляду на нульову активність ферменту GAA тяжкий перебіг захворювання і смерть хворого у ранньому віці ці мутації слід вважати високопатогенними. В інших хворих виявлено описані раніше мутації (їх спостерігають досить рідко, за ювенільного та пізнього інфантильного типів захворювання) [20–23] (табл. 2). Швидше за все, виявлено спектр мутацій, притаманний некласичному типу ІФХП.
У 3 хворих на ХФ упродовж трьох років застосовували ФЗТ. Своєчасне призначення лікування при ІФХП (клінічне спостереження 3) дозволило уникнути ранньої смерті дитини (ще 2 хворі, яким ФЗТ не призначали, померли у віці до 7 міс.). З іншого боку, у 2 хворих за атипової форми ІФХП діагноз встановлений запізно. В однієї дитини ФЗТ довелося розпочинати у реанімаційному відділенні. Однак навіть пізній початок лікування дозволив зберегти життя хворих, відновити рухову активність: в одного пацієнта майже повністю відновилася здатність самостійно ходити, відзначено покращення функції дихальної та серцево-судинної систем. Невелика кількість спостережень не дозволяє зробити вірогідні висновки щодо ефективності ФЗТ, проте попередні результати нашого дослідження є досить оптимістичними і збігаються з даними інших дослідників [19, 24].
Аналіз частоти виявлення ХП у різних географічних регіонах та етнічних групах свідчить, що частіше це захворювання діагностують у дорослих осіб, у яких також описаний його асимптомний перебіг [25]. Крім того, клінічна картина ХП у дітей відрізняється від такої у дорослих пацієнтів. Саме тому при побудові діагностичного алгоритму важливо враховувати вік пацієнтів, особливості мультисистемних клінічних проявів, біохімічний фенотип, дані інструментальних досліджень — це необхідно для формування селективних груп пацієнтів із підо–зрою на ХП для проведення подальшої лабораторної діагностики методом СКК.
Важливим при діагностиці ХП у дітей є аналіз гетерогенної клінічної картини: основними симптомами у дітей віком до 6 місяців є порушення з боку серця (гіпертрофічна кардіоміопатія лівого шлуночка) у поєднанні з міопатичним синдромом і специфічним біохімічним фенотипом (підвищення рівня АЛТ, АСТ, КФК). Беручи до уваги наявність некласичного варіанта ІФХП, слід пам’ятати, що основним у такій ситуації є міопатичний синдром у поєднанні з біохімічними особливостями. На нашу думку, запропонований діагностичний алгоритм сприятиме оптимізації ранньої діагностики ХП і своєчасному призначенню ФЗТ (рис. 3).
За результатами нашого дослідження та даними інших авторів [2, 15, 16], діагностика ХП є надзвичайно складною, у більшості спостережень діагноз встановлюють запізно.
Висновки
Клінічна картина ХП у дітей характеризується мультисистемним ураженням і різноманітними клінічними формами, за відсутності специфічного лікування призводить до ранньої смерті хворих.
Основними проявами ХП у дітей є гіпертрофічна кардіоміопатія, міопатичний синдром у поєднанні зі специфічним біохімічним фенотипом.
Метод ферментодіагностики в СКК у пацієнтів дитячого віку з певними клінічними синдромами в рамках селективного скринінгу дозволить підвищити ефективність ранньої діагностики та лікування цієї орфанної хвороби.
Вдячність. Автори висловлюють щиру подяку лікарям-лаборантам медико-генетичної лабораторії НДСЛ «Охматдит», лабораторії Archimed Life Science GmbH (Відень, Австрія), компанії «Джензайм» у складі «Санофі», лікарю-педіатру Н.І. Сінчук, лікарю-неврологу І.В. Македонській, а також усім лікарям, які направляли хворих на консультацію до Центру орфанних захворювань.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Lysosomal storage disorders. A practical guide / ed. by A. Mehta, B. Winchester. — Hoboken: Wiley-Blackwell, 2012. — 101 p.
2. Kishnani P.S. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysio–logy, and management / P.S. Kishnani, A.A. Beckemeyer, N.J. Mendelsohn // Am. J. Med. Genet. C: Semin. Med. Genet. — 2012. — Vol. 160C, № 1. — P. 1-7. doi: 10.1002/ajmg.c.31324.
3. Diagnosis of late-onset Pompe disease and other muscle di–sorders by next-generation sequencing / S. Lévesque, C. Auray-Blais, E. Gravel et al. // Orphanet. J. Rare Dis. — 2016. — Vol. 11. — P. 8-9. doi: 10.1186/s13023-016-0390-6.
4. A large-scale nationwide newborn screening program for Pompe disease in Taiwan: towards effective diagnosis and treatment / –C.-F. Yang, H.-C. Liu, T.-R. Hsu et al. // Am. J. Med. Genet. A. — 2014. — Vol. 164, № 1. — P. 54-61. doi: 10.1002/ajmg.a.36197.
5. Pompe J.-C. Over idiopatische hypertrophie van het hart / J.-C. Pompe // Ned. Tijdshr. Geneeskd. — 1932. — Vol. 76. —
P. 304.
6. Pompe disease diagnosis and management guideline / P.S. Kishnani, R.D. Steiner, D. Bali et al. // Genet. Med. — 2006. — Vol. 8. — P. 267-288. PMID: 16702877.
7. Infantile-onset pompe disease natural history study G: a retrospective, multinational, multicenter study on the natural history of infantile-onset pompe disease / P.S. Kishnani, W.L. Hwu, H. Mandel et al. // J. Pediatr. — 2006. — Vol. 148. — P. 671-676. doi: 10.109701.gim.0000218152.87434.f3.
8. Survival and associated factors in 268 adults with pompe di–seаse prior to treatment with enzame replacement therapy / D. Gungor, J.M. de Vries, W.C. Hop et al. // Orphanet. J. Rare Dis. — 2011. — Vol. 6. — P. 34. doi: 10.1186/1750-1172-6-34.
9. van der Ploeg A.T. Pompe’s disease / A.T. van der Ploeg, A.J. Reuser // Lancet. — 2008. — Vol. 372, № 9646. — P. 1342-1353. doi: 10.1016/S0140-6736(08)61555-X.
10. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus mee–ting / B. Winchester, D. Bali, O.A. Bodamer et al. // Mol. Genet. Metab. — 2008. — Vol. 93, № 3. — P. 275-281. doi: 10.1016/j.ymgme.2007.09.006.
11. Diagnostic efficacy of the fluorometric determination of enzyme activity for Pompe disease from dried blood specimens compared with lymphocytes-possibility for newborn screening / Z. Lukacs, P. Nieves Cobos, E. Mengel et al. // J. Inherit. Metab. Dis. — 2010. — Vol. 33, № 1. — P. 43-50. doi: 10.1007/s10545-009-9003-z.
12. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease / P.S. Kishnani, D. Corzo, N.D. Leslie et al. // Pediatr. Res. — 2009. — Vol. 66. — P. 329-335. doi: 10.1203/PDR.0b013e3181b24e94
13. Rapіd dіagnоstіc testіng prоcedures fоr lysоsоmal stоrage dіsоrders: alpha-glucоsіdase and betagalactоsіdase assays оn drіed blооd spоts / N. Gasparоttо, R. Tоmanіn, A.C. Frіgо et al. // Clіn. Chіm. Acta. — 2009. — Vol. 402, № 1-2. — P. 38-41. doi: 10.1016/j.cca.2008.12.006.
14. Gaucher and Nіemann-Pіck dіseases. Enzymatіc dіagnоsіs іn drіed blооd spоts оn fіlter paper: retrоspectіve dіagnоses іn newbоrn-screenіng cards / N.A. Chamоles, M.B. Blancо, D. Gaggіоlі, C. Casentіnі // Clіn. Chіm. Act. — 2002. — Vol. 317. — P. 191-197.
15. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry / P.S. Kishnani, H.M. Amartino, C. Lindberg et al. // Am. J. Med. Genet. A. — 2013. — Vol. 161A, № 10. — P. 2431-2443. doi: 10.1002/ajmg.a.36110.
16. Childhood Pompe disease: clinical spectrum and genotype in 31 patients / C.I. van Capelle, J.C. van der Meijden, J.M.P. van den Hout et al. // Orphanet. J. Rare Dis. — 2016. — Vol. 11, № 1. — P. 65. doi: 10.1186/s13023-016-0442-y.
17. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations / A. Herzog, R. Hartung, A.J.J. Reuser et al. // Orphanet. J. Rare Dis. — 2012. — Vol. 7. — P. 35. doi: 10.1186/1750-1172-7-35.
18. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia / T. Kyriakides, C. Angelini, J. Schaefer et al. // Eur. J. Neurol. 2010. — Vol. 17, № 6. — P. 767-773. doi: 10.1111/j.1468-1331.2010.03012.x.
19. Yin-Hsiu Chien. Pompe disease: early diagnosis and early treatment make a difference / Yin-Hsiu Chien, Wuh-Liang Hwu, Ni-Chung Lee // Pediatr. Neonatol. — 2013. — Vol. 54, № 4. — P. 219-227. doi: 10.1016/j.pedneo.2013.03.009.
20. Juvenile-onset glycogen storage disease type II with novel mutations in acid alpha-glucosidase gene / C.W. Lam, Y.P. Yuen, K.Y. Chan et al. // Neurology. — 2003. — Vol. 60, № 4. — P. 715-717. PMID: 12601120.
21. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease / M.G. Pittis, M. Donnarumma, A.L. Montalvo et al. // Hum. Mutat. — 2008. — Vol. 1007, № 29. — P. 27-36. doi: 10.1002/humu.20753.
22. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating / M. Kroos, R.J. Pomponio, L. van Vliet et al. // Hum. Mutat. — 2008. — Vol. 1007, № 29. — P. 13-26. doi: 10.1002/humu.20745.
23. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II / M.M. Hermans, D. van Leenen, M.A. Kroos et al. // Hum. Mutat. — 2004. — Vol. 23, № 1. — P. 47-56. doi: 10.1002/humu.10286.
24. Phenotypical variation within 22 families with Pompe disease / S.C.A. Wens, C.M. van Gelder, M.E. Kruijshaar et al. // Orphanet. J. Rare Dis. — 2013. — Vol. 8. — P. 182. doi: 10.1186/1750-1172-8-182.
25. Targeted screening for the detection of Pompe disease in patients with unclassified limb-girdle muscular dystrophy or asymptomatic hyperCKemia using dried blood: of Spanish cohort / E. Gutiérrez-Rivas, J. Bautista, J.J. Vílchez et al. // Neuromusc. Disord. — 2015. — Vol. 25, № 7. — P. 548-553. doi: 10.1016/j.nmd.2015.04.008.

/70-1.jpg )
/71-1.jpg )
/72-1.jpg )
/11-1.jpg )