По современным представлениям, терапевтическое действие амантадина связано с блокадой глутаматных NMDA (N-methyl-D-aspartat) рецепторов и ослаблением возбуждающих кортикостриарных влияний на холинергические нейроны стриатума [2–4]. Показано, что амантадин обладает способностью стимулировать высвобождение накопившегося дофамина из пресинаптических терминалей и блокировать обратный его захват. В частности, в клинико-экспериментальных исследованиях установлено, что благоприятный эффект амантадина связан со способностью уменьшать дисбаланс между дофаминовой и глутаматной нейромедиаторной системой на всех этапах клинического развития болезни Паркинсона (БП) [10, 13, 14, 18]. Так, амантадин широко применяется как на ранних стадиях заболевания с целью отсрочить назначение леводопасодержащих препаратов, так и на более поздних, для потенцирования действия других антипаркинсонических средств и влияния на осложнения леводопатерапии, таких как дискинезии и моторные флюктуации [3, 4, 21, 28, 29].
Обосновано также положение о том, что и другие заболевания ЦНС, сопровождающиеся нейрональной потерей вследствие глутаматной эксайтотоксичности, могут быть мишенью для лечения амантадином. Этот перечень наряду с болезнью Паркинсона включает церебральную ишемию, посттравматическую энцефалопатию, эпилепсию, когнитивные нарушения, нейропатические боли и т.д. [2, 5–7, 28]. Убедительно показан нейропротекторный и цитопротекторный эффект амантадина на моделях паркинсонизма, деменции, инсульта [7, 8, 10, 11, 24, 30].
Структура амантадина в настоящее время достаточно глубоко изучена. Считают, что амантадин — это противовирусный и одновременно противопаркинсонический препарат, по своей химической природе представляющий трициклический аминоадамантан, производное адамантана — 1-аминоадамантана гидро–хлорид, который хорошо растворим в воде, легко абсорбируется после приема внутрь. Пиковая концентрация препарата в крови наступает спустя 2–4 ч после приема таблетированной формы, а период полужизни в плазме крови составляет от 10 до 24 ч. Препарат экскретируется с мочой, практически не метаболизируясь в организме [4, 12, 18].
Амантадин был предложен в начале 1960-х годов в качестве противовирусного средства, эффективного в отношении вирусов гриппа типа А2. Спустя несколько лет случайно была установлена его эффективность при паркинсонизме, когда уменьшение симптомов заболевания неожиданно было обнаружено у 58-летней женщины, страдавшей БП и принимавшей амантадин для профилактики гриппа. Вскоре после этого было проведено специально организованное клиническое исследование, включавшее 163 больных БП, в котором было четко показано улучшение состояния у 66 % пациентов, принимавших амантадин (Schwab R.S. et al., 1969).
На практике амантадиновые производные в настоящее время наиболее часто назначают в качестве монотерапии на начальных стадиях развития БП, в дополнение к леводопе или агонисту дофаминовых рецепторов — на более поздних этапах заболевания. Оптимальная стандартная доза амантадина — 200–300 мг/сут в 3 приема, стабильная концентрация препарата в крови достигается в течение 4–7 дней. Амантадин обычно хорошо переносится пациентами разных возрастных групп, хотя после 70–75 лет его следует принимать с осторожностью. Побочные эффекты редки и могут проявляться в виде отеков (чаще голеней и стоп), сухости во рту, «мраморности» кожных покровов, нарушений сна, эпизодов возбуждения и галлюцинаций.
Амантадин оказывает умеренный эффект в отношении всех основных клинических проявлений паркинсонизма, что наблюдается почти у 50 % пациентов [1, 3, 4, 28, 29]. Улучшение касается в основном гипокинезии, ригидности, тремора. На развернутых стадиях БП амантадин при добавлении к леводопе может усиливать ее действие и в то же время уменьшать выраженность двигательных осложнений проводимой леводопатерапии [20, 23]. Доказано свойство амантадина влиять на выраженность леводопаиндуцированных дискинезий, инвалидизирующих пациентов.
В настоящее время амантадин является единственным препаратом, у которого антидискинетическая активность при БП имеет убедительную доказательную базу [9, 15, 16, 20].
Благоприятное влияние амантадина на лекарственные дискинезии у пациентов с БП подтверждено и в эксперименте на обезьянах с токсической моделью паркинсонизма [3, 10].
Получено достаточное количество результатов двойных слепых контролируемых исследований, показавших, что амантадин способен уменьшать тяжесть и длительность лекарственных дискинезий при БП без влияния на противопаркинсонические эффекты леводопы и ухудшения общих двигательных функций [16, 17, 23, 33–35].
Во всех указанных работах продолжительность активного периода лечения составляла от 2 до 4 недель, а результаты исследования длительной антидискинетической активности амантадина оставались неоднозначными. Так, по данным L.V. Metman et al., поддержание хорошего терапевтического эффекта амантадина имело место в течение года у 13 из 17 пациентов [25]. E. Wolf et al. провели рандомизированное плацебо-контролируемое исследование в параллельных группах, включавшее 32 больных БП, длительно (в среднем 4,8 года) принимавших стабильную дозу амантадина [38]. У этих пациентов при переводе на плацебо уже через 3 недели наблюдалось достоверное увеличение выраженности и длительности леводопаиндуцированых дискинезий согласно оценке по IV части шкалы UPDRS. Эти данные подтверждают долговременную антидискинетическую эффективность амантадина на развернутой стадии БП [27].
Представляет значительный интерес опубликованный крупный метаанализ результатов контролируемых исследований антагонистов NMDA-рецепторов у пациентов с БП, осложненной развитием леводопаиндуцированных дискинезий, который включал 11 плацебо-контролируемых исследований с участием 253 больных [17]. В итоге было сделано заключение о достоверном влиянии амантадина на снижение дискинезии пика дозы и уменьшение общей тяжести симптомов по III и IV частям шкалы UPDRS.
С тех пор антагонист глутаматных NMDA-рецепторов амантадин имеет все основания активно использоваться у больных БП для коррекции лекарственных дискинезий.
Наряду с антидискинетическим эффектом доказано нейропротективное действие амантадина, которое связано не только с блокадой NMDA-рецепторов, но и с влиянием на процессы микроглиального воспаления, а также с повышением выработки нейротрофических факторов астроглией. Так, в исследовании с МФТП-индуцированным паркинсонизмом на культуре клеток среднего мозга амантадин в 4–5 раз снижал токсический эффект МФТ (1-метил-4-фенилпиридиний) и липосахарида, вызывающих гибель дофаминергических нейронов. Более того, амантадин предотвращал активацию микроглии и продукцию противовоспалительных факторов, таких как фактор некроза опухоли, простагландины, оксид азота, что свидетельствует о наличии у него противовоспалительного эффекта [1, 30]. Известно, что активированная микроглия является важнейшим звеном в патогенезе нейродегенеративного процесса, усиливая формирование протофибрилл α-синуклеина и их агрегацию. Эти результаты согласуются с экспериментальными данными об уменьшении активации макрофагов на фоне применения амантадина, сопровождающемся повышением экспрессии нейротрофического фактора глиальных клеток [32]. Этот эффект был подтвержден в исследованиях, показавших влияние амантадина на повышение экспрессии фактора роста нервов BDNF (мозгового нейротрофического фактора) и GDNF (глиального нейротрофического фактора) в корковых нейронах крыс и в клетках глиомы [11, 32]. Именно нейропротективным эффектом амантадина объясняются результаты ретроспективного исследования R.J. Uitti et al., доказавшего, что длительный прием амантадина сопровождается увеличением продолжительности жизни [36].
Антидискинетический эффект амантадина был продемонстрирован также в целом ряде плацебо-контролируемых исследований, в которых установлено значительное снижение выраженности и продолжительности дискинезий без уменьшения периода включения. Прием амантадина приводил к снижению дискинезий пика дозы в среднем у 40–60 % больных [8, 23]. В другом многоцентровом плацебо-контролируемом перекрестном исследовании с использованием амантадина в суточной дозе 300 мг на протяжении 1 мес. было показано, что препарат уменьшает выраженность дискинезий по шкале UPDRS-IV на 64,7 % [34]. Путем мультивариационного анализа было установлено, что наилучший эффект амантадин демонстрирует при назначении пациентам с более поздним началом заболевания и ранее применявшим АДР [22, 33]. Некоторые авторы связывают реализацию антидискинетического эффекта амантадина с действием на уровне STN, так как препарат оказался неэффективен в отношении дискинезий, развившихся после субталамотомии [26]. Результаты Кокрановского обзора, опубликованного в 2003 г., свидетельствуют о безопасности и эффективности амантадина в отношении леводопаиндуцированных дискинезий [15]. В связи с этим в настоящее время амантадин включен в европейские и американские рекомендации как препарат первой линий при лечении дискинезий. Рекомендуемая суточная доза препарата может варьировать в диапазоне от 200 до 400 мг.
В августе 2017 г. FDA утвердило амантадина гидро–хлорид (GOCOVRI (ADS-5102)) с пролонгированным высвобождением как препарат для улучшения контроля дискинезий, вызванных леводопой. В опубликованных результатах многоцентровых исследований EASED, EASELID и EASELID 3 продолжительностью 8 недель при использовании препарата в дозах 260, 340 и 420 мг/сут была продемонстрирована эффективность всех указанных дозировок (рис. 1). При применении препарата в дозе 340 мг отмечалось снижение баллов по шкале UDysRS (Unified Dyskinesia Rating Scale) на 27 % (р < 0,005), увеличивался период включения без тяжелых инвалидизирующих дискинезий [31]. Препарат назначался однократно вечером перед сном. Пик его концентрации достигался через 12–14 часов после приема, что обеспечивало максимальный эффект в отношении дискинезии в утренние и дневные часы и, что очень важно, не ухудшало сон. Амантадин с пролонгированным высвобождением (GOCOVRI) показал стабильный антидискинетический эффект в течение 6 месяцев.
/99-1.jpg)
Еще одно важнейшее свойство амантадина заключается в его влиянии на когнитивные функции. Ранее считалось, что болезнь Паркинсона необязательно сопровождается когнитивной дисфункцией, однако сейчас появляется все больше доказательств частого развития деменции при этом заболевании. Количество пациентов с БП, у которых при прогрессировании болезни развивается деменция, составляет 40–60 %. Отмечены такие факторы риска, как возраст на момент обследования, возраст к началу заболевания, длительность течения, акинетико-ригидная форма заболевания, депрессия и атипичные для БП симптомы. Наличие БП в семейном анамнезе повышает склонность к деменции, хотя аллель Аро Е4 не является фактором риска для развития деменции при БП [1, 4]. У пациентов пожилого возраста с тяжелыми симптомами БП риск развития деменции приблизительно в 10 раз больше, а деменция повышает риск гибели пациентов, страдающих БП [4, 7]. В связи с этим патогенетически значимое лечение, позволяющее задержать развитие или уменьшить тяжесть деменции, может способствовать увеличению продолжительности жизни пациентов с БП.
Нарушение функциональной активности мозга, обнаруживаемое при деменции, сопровождается устойчивым повышением уровня глутамата и/или развивающейся гиперсенситивностью NMDA-рецепторов, что приводит к усилению поступления кальция в нейроны, нарушению нейронального гомеостаза и последующей дегенерации нервных клеток [3, 28, 29]. В дальнейшем чрезмерная глутаматергическая стимуляция гип–покампа вызывает функциональные нарушения в проводящих путях, играющих важнейшую роль в процессе обучения и запоминания. Более того, глутамат активирует N-метил-D-аспартатные рецепторы, участвующие в формировании центральных механизмов, отвечающих за обучение и память [7, 24].
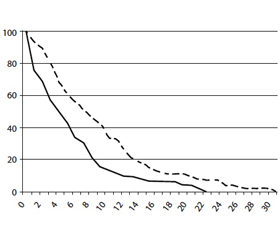
Роль блокаторов NMDA-рецепторов в патогенетической терапии когнитивных нарушений при БП показана в крупномасштабном исследовании связи между приемом амантадина и началом деменции при БП [19]. Целью этого исследования было сравнение частоты развития деменции у пациентов, страдающих БП, получавших лечение амантадином (группа АМ) и никогда его не получавших (группа NоАМ). С помощью метода Kaplan-Meier выполнена оценка кривых выживаемости для пациентов из группы АМ и группы NоАМ, при этом в качестве конечной точки принято появление симптомов деменции. Популяция исследования состояла из 539 пациентов (возраст 69,5 ± 9,9 года; длительность БП 9,2 ± 6,0 года; 263 пациента (44 %) получали лечение амантадином). Конечная точка деменции была достигнута 116 пациентами (20 %). Длительность течения БП до развития деменции оказалась значимо больше в группе АМ (9,1 ± 5,7 года) по сравнению с группой NоАМ (5,9 ± 4,6 года, Р = 0,006). Установлено, что продолжительность лечения амантадином имеет положительную связь с длительностью течения БП до развития деменции (Р = 0,0001). Анализ выживаемости с учетом деменции в качестве конечной точки показал более медленное снижение умственных способностей в группе АМ. Показатели мини-теста оценки психического состояния (ММSЕ) в группе АМ оказались существенно выше, чем в группе NоАМ (Р = 0,010). Возраст появления признаков БП также оказывал значимое влияние на продолжительность течения БП до развития деменции. В связи с этим сделан убедительный вывод о том, что применение амантадина позволяет отстрочить появление признаков деменции у страдающих БП пациентов, а также уменьшить ее тяжесть (рис. 2).
/100-1.jpg)
Амантадин в лечении нейролептических экстрапирамидных синдромов
Известно, что нейролептики могут вызывать практически весь спектр экстрапирамидных расстройств: паркинсонизм, дистонию, тремор, хорею, апатию, тики, миоклонию и др. [4]. В большинстве случаев экстрапирамидные синдромы, возникшие на фоне приема того или иного препарата, после его отмены постепенно регрессируют, но некоторые разновидности нейролептических дискинезий и дистоний носят стойкий характер и персистируют даже после отмены вызвавшего их препарата. Нейролептики, как блокаторы Д2-рецепторов, вызывают так называемые ранние и поздние экстрапирамидные синдромы. Ранние возникают в течение первых дней или недель после приема нейролептика или на фоне увеличения его дозы. Они обычно регрессируют вскоре после отмены препарата или при переводе больного на атипичный нейролептик. Поздние возникают вследствие длительного, в течение нескольких месяцев или лет, приема нейролептика, иногда вскоре после его отмены, имеют стойкий или даже необратимый характер. В настоящее время широко обсуждается вопрос о терапевтической эффективности препаратов амантадина в купировании экстрапирамидных нейролептических синдромов [6]. Показано, что амантадин в дозе 200 мг/сутки терапевтически эквивалентен 8 мг/сутки тригексифенидила, эффективен при поздней дискинезии, не уменьшает антипсихотический эффект нейролептиков, не вызывает когнитивных нарушений, оказывает нейропротекторный эффект.
Применение антагонистов глутаматных рецепторов (амантадина) при хронической мозговой сосудистой недостаточности с явлениями сосудистого паркинсонизма
Теоретическим обоснованием применения амантадина при острой недостаточности мозгового кровообращения и его последствиях является возникновение энергетического дефицита вследствие ишемии, который является причиной деполяризации мембран и высвобождения эксайтотоксического глутамата, вызывающего вазоконстрикцию, повышение проницаемости стенок сосудов. Избыточное внутриклеточное накопление Са2+ в ишемизированных нейронах и переход его в активную форму вызывает активацию клеточных ферментов, обусловливающих образование NO, свободных радикалов, разрушение внутриклеточных белков, фосфолипидов, нуклеиновых кислот и гибель клетки [3, 4, 28].
Вот почему назначение препаратов амантадинового ряда при хронической ишемии мозга и в процессах постинсультной реабилитации может способствовать нейропротекции поврежденных ишемизированных нейронов. Отмечают, что на фоне лечения амантадином больных в реабилитационном периоде после инсульта происходило уменьшение неврологического дефицита по шкале NIHSS на 39,1 %; при этом хороший эффект не зависел от возраста больных и подтипа ишемического инсульта [5]. В контрольной группе на 10-й день госпитализации оценка по шкале NIHSS снизилась только на 24,4 % (Р < 0,005).
Таким образом, фармакотерапевтическая эффективность амантадина подтверждена во многих исследованиях. Учитывая, что амантадин, как блокатор глутаматных NMDA-рецепторов, оказывает патогенетическое действие на эксайтотоксический каскад, участвующий в патогенезе основных заболеваний ЦНС, значение этого класса препаратов в лекарственной терапии трудно переоценить. Амантадин оказался эффективным и при некоторых других экстрапирамидных синдромах, имеющих дофаминергические механизмы развития, например при хорее Гентингтона [37]. Следует полагать, что возможности применения амантадина в неврологии будут еще более расширяться в связи со спектром его нейромедиаторной активности при целом ряде патологических состояний, таких как реабилитация после черепно-мозговой травмы, воспалительных и вирусных заболеваниях ЦНС, полинейропатиях и т.д.
Конфликт интересов. Не заявлен.
Список литературы
1. Иллариошкин С.Н., Федотова Е.Ю., Селиверстов Ю.А. Современные высокотехнологичные методы лечения болезни Паркинсона // Болезни мозга: медицинские и социальные аспекты / Под ред. Е.И. Гусева, А.Б. Гехт. — М.: Буки-Веди, 2016. — С. 543-552.
2. Карабань И.Н. Применение блокатора глутаматных рецепторов амантадина в неврологии // Международный неврологический журнал. — 2012. — № 2 (48). — С. 195-201.
3. Крыжановский Г.Н., Карабань И.Н., Магаева С.В., Кучеряну В.Г., Карабань Н.В. Болезнь Паркинсона. — М.: Медицина, 2002. — 336 с.
4. Левин О.С., Федорова Н.В. Болезнь Паркинсона. — М.: Медпрессинформ, 2017. — 315 с.
5. Румянцева С.А., Беневольская Н.Г. Новые направления в патогенетической терапии инсульта // Атмосфера. Нервные болезни: журнал для практических врачей. — 2006. — № 4. — С. 29-34.
6. Федорова Н.В., Ветохина Т.Н. Диагностика и лечение нейролептических экстрапирамидных синдромов: Учебно-методическое пособие. — М.: РАМПО, 2006. — 36 с.
7. Aarsland D., Ballard C., Walker Z. Memantine in patients with Parkinson’s disease, dementia or dementia with Lewy bodies // Lancet Neurol. — 2009. — V. 8. — P. 613-618.
8. Ahmed I., Bose S.K., Pavese N., Ramlackhansingh A., Turkheimer F., Hotton G., Hammers A., Brooks D.J. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias // Brain. — 2011. — 134 (Pt. 4). — 979-986.
9. Bezard E., Brotchie J.M., Gross C.E. Pathophysiology of levodopainduced dyskinesia: potential for new therapies // Nat. Rev. Neurosci. — 2001. — 2. — 577-588.
10. Blanchet P.J., Konitsiotis S., Chase T.N. Amantadine reduces levodopainduced dyskinesias in parkinsonian monkeys // Mov. Disord. — 1998. — 13. — 798-802.
11. Caumont A.S., Octave J.N., Hermans E. Amantadine and memantine induce the expression of the glial cell line-derived neurotrophic factor in C6 glioma cells // Neurosci Lett. — 2006. — 394. — 196-201.
12. Cersósimo M.G., Micheli F.E. Antiglutamatergic drugs in the treatment of Parkinson’s disease / Koller C., Melamed E., editors // Handbook of clinical neurology. V. 84: Parkinson’s disease and related disorders. Part II. — N.Y.: Elsevier, 2007. — Р. 127-136.
13. Chase T.N., Bibbiani F., Oh J.D. Striatal glutamatergic mecha–nisms and extrapyramidal movement disorders // Neurotox. Res. — 2003. — 5 (1–2). — 139-146.
14. Chase T.N., Engber T.M., Mouradian M.M. Contribution of dopaminergic and glutamatergic mechanisms to the pathogenesis of motor response complications in Parkinson’s disease // Adv. Neurol. — 1996. — 69. — 497-501.
15. Crosby N.J., Deane K.H., Clarke C.E. Amantadine for dyskinesia in Parkinson’s disease // Cochrane Database Syst. Rev. — 2003. — 2. — CD003467.
16. Da Silva-Junior F.P., Braga-Neto P., Sueli Monte F.,
De Bruin V.M. Amantadine reduces the duration of levodopa-induced dyskinesia: a randomized, double-blind, placebo-controlled study // Parkinsonism Relat. Disord. — 2005. — 11. — 449-452.
17. Elahi B., Phielipp N., Chen R. N-methyl-D-aspartate antagonists in levodopa induced dyskinesia: a meta-analysis // Can. J. Neurol. Sci. — 2012. — 39. — 465-472.
18. Halliday G. The progression of pathology in Parkinson’s dise–ase // Ann. N.Y. Acad. Sci. — 2010. — V. 1184. — Р. 188-195.
19. Ilzenberg R., Bonuccelli U., Schechtman E., Miniowich A., Strugatsky R., Ceravolo R., Logi C., Rossi C., Klein C., Rabey J.M. Association between amantadine and the onset of dementia in Parkinson’s disease // Mov. Disord. — 2006. — 21. — 1375-1379.
20. Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations // Mov. Disord. — 2005. — 20 (Suppl. 11). — S11-S16.
21. Jankovic J., Stacy M. Medical management of levodopa-–associated motor complications in patients with Parkinson’s disease //CNS Drugs. — 2007. — 21. — 677-692.
22. Kaplan N., Vituri A., Korczyn A.D., Cohen O.S., Inzelberg R., Yahalom G., Kozlova E., Milgrom R., Laitman Y, Friedman E., Rosset S., Hassin-Baer S. Sequence variants in SLC6A3, DRD2, and BDNF genes and time to levodopa-induced dyskinesias in Parkinson’s disease // J. Mol. Neurosci. — 2014. — 53. — 183-188.
23. Luginger E., Wenning G.K., Boesch S., Poewe W. Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease // Mov. Disord. — 2000. — 15. — 873-878.
24. Melki R. Role of different alpha-synuclein strains in synucleinopa-thies, similarities with other neurodegenerative diseases // J. Parkinsons Dis. — 2015. — 5 (2). — 217-227.
25. Metman L.V., DelDotto P., LePoole K. et al. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study // Arch. Neurol. — 1999. — 56. — 1383-1386.
26. Merello M., Perez-Lloret S., Antico J., Obeso J.A. Dyskinesias induced by subthalamotomy in Parkinson’s disease are unresponsive to amantadine // J. Neurol. Neurosurg. Psychiatry. — 2006. — 77 (2). — 172-174.
27. Nutt J.G., Chung K.A., Holford N.H. Dyskinesia and the antiparkinsonian response always temporally coincide: a retrospective study // Neurology. — 2010. — V. 74 (15). — 1191-1197.
28. Olanow C.W. The scientific basis for the current treatment of Parkinson’s disease // Annu Rev. Med. — 2004. — 55. — 41-60.
29. Olanow C.W., Stern M.B., Sethi K. The scientific and clinical basis for the treatment of Parkinson’s disease // Neurology. — 2009. — V. 72, 21 (Suppl. 4). — Р. 149.
30. Ossola B., Schendzielorz N., Chen S.H., Bird G.S., Tuominen R.K., Männistö P.T., Hong J.S. Amantadine protects dopamine neurons by a dual action. — reducing activation of microglia and inducing expression of GNDF in astroglia // Neuropharmacology. — 2011. — 61 (4). — 574-582.
31. Pahwa R., Tanner C.M., Hauser R.A., Sethi K., Isaacson S., Truong D., Struck L., Ruby A.E., McClure N.L., Went G.T., Stempi–en M.J. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study) // Mov. Disord. — 2015. — 30 (6). — 788-795.
32. Roman A., Rogoz Z., Kubera M. et al. Concominent administration of fluoxetine and amantadine modulates the activity of peritoneal macrophages of rats subjected to a forced swimming test // Pharmacol. Rep. — 2009. — 61. — 1069-1077.
33. Sawada H., Oeda T., Kuno S. et al. Amantadine Study Group. Amantadine for dyskinesias in Parkinson’s disease: a randomized controlled trial // PloS One. — 2010. — 5 (12). — e 15298.
34. Stocchi F., Rascol O., Destee A., Hattori N., Hauser R.A., Lang A.E., Poewe W., Stacy M., Tolosa E., Gao H., Nagel J., Merschhemke M., Graf A., Kenney C., Trenkwalder C. AFQ056 in Parkinson patients with levodopa-induced dyskinesia. — 13-week, randomized, dose-finding study // Mov. Disord. — 2013. — 28 (13). — 1838-1846.
35. Thomas A., lacono D., Luciano A.L., Armellino K., Di lorio A., Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease // J. Neurol. Neurosurg. Psychiatry. — 2004. — 75. — 141-143.
36. Uitti R.J., Rajput A.H., Ahlskog J.E., Offord K.P., Schroeder D.R., Ho M.M., Prasad M., Rajput A., Basran P. Amantadine treatment as an independent predictor of improved survival in Parkinson’s disease // Neurology. — 1996. — 46. — 1551-1556.
37. Verhagen Metman L., Morris M.J., Farmer C., Gillespie M., Mosby K., Wuu J., Chase T.N. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine // Neuro–logy. — 2002. — 59. — 694-699.
38. Wolf E., Seppi K., Katzenschlager R., Hochschorner G., Rans-mayr G., Schwingenschuh P., Ott E., Kloiber I., Haubenberger D., Auff E., Poewe W. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease // Mov. Disord. — 2010. — 25. — 1357-1363.