Вступ
Вірус варицела-зостер (Varicella Zoster Virus — VZV) відомий як збудник вітряної віспи при первинному інфікуванні й оперізуючого герпесу — при реактивації вірусу з латентного стану. Останнім часом накопичено багато даних щодо альтернативних клінічних проявів реактивованої інфекції, викликаної VZV, завдяки чому значно розширилися уявлення про гетерогенність маніфестації VZV-індукованих уражень нервової системи. Встановлено, що VZV може викликати серозний лімфоцитарний менінгіт, мультифокальний лейкоенцефаліт, стовбуровий енцефаліт, церебеліт, різні форми мієлітів, ураження краніальних нервів, брахіоплексопатію і так званий абдомінальний герпес, які можуть розвиватися як у хронологічному зв’язку з вітряною віспою або оперізуючим герпесом, так і без попереднього періоду герпетичних висипань на шкірі [4]. Васкулопатії церебральних судин VZV-етіології на сьогодні розглядаються як поширена в популяції, однак усе ще недооцінена тяжка патологія нервової системи, що може стати причиною інвалідності або передчасної смерті пацієнта [1, 5]. У даному науковому огляді проведено аналіз доступних даних щодо патогенезу, клінічних проявів, діагностики, лікування й профілактики VZV-васкулопатій церебральних судин у людей.
Патогенез
Вірус потрапляє до організму людини аерозольним шляхом і використовує для проникнення в епітеліоцити рецептор, який за природою є ферментом, що руйнує інсулін. Клінічна картина первинної інфекції позначається терміном «вітряна віспа».
Як відомо, після епізоду первинної інфекції VZV довічно зберігається в латентному стані в сенсорних гангліях периферичної нервової системи, включаючи краніальні, паравертебральні й вегетативні нервові вузли. Здатність VZV до латентності в чутливих гангліях продемонстрована в класичному дослідженні F.O. Bastian зі співавт. [13]. Пізніше E.R. Richter зі співавт. вивчили розподіл Herpes simplex virus 1 (HSV1) і VZV у сенсорних і вегетативних гангліях голови і шиї, вивчаючи фіксовані в парафіні зразки тканини, отримані при автопсії. HSV1 і VZV були ідентифіковані в 18 із 58 (31 %) і 16 з 58 (28 %) трійчастих гангліїв, 23 із 58 (40 %) і 11 із 58 (19 %) крилопіднебінних, 25 із 60 (42 %) і 14 із 60 (23 %) циліарних, 25 із 48 (52 %) і 11 із 48 (23 %) колінчастих, 15 із 50 (30 %) і 8 із 50 (16 %) вушних, 14 із 47 (30 %) і 4 із 47 (9 %) підщелепних, 18 із 58 (31 %) і 10 із 58 (17 %) верхніх шийних, 12 із 36 (33 %) і 1 із 36 (3 %) нодозних гангліїв відповідно [61]. Наразі теорія латентності вірусу в периферичних сенсорних нервових гангліях доопрацьована й обґрунтована Ізраелем Стейнером. Вона базується на даних імуногістохімічного аналізу та нозерн-блотингу, що дозволяють ідентифікувати вірус всередині нервових вузлів протягом прихованого періоду перебігу інфекції (рис. 1) [70].
/100-1.jpg)
У сенсорних нервових гангліях периферичної нервової системи вірус, на відміну від HSV1 і HSV2, перебуває не в нейронах, а в сателітних клітинах у стані обмеженої експресії геному під тиском імунної відповіді з боку резидентних природних кілерів [7, 12], природних кілерних Т-лімфоцитів [27] (рис. 2) і CD8+ цитотоксичних Т-лімфоцитів [69]. Останні обробляють інфіковані вірусом клітини ганглія альфа-, бета- і лямбда-інтерферонами, що чинять віростатичну дію й перешкоджають відновленню повноцінної репродукції вірусу. Це так званий інтрагангліонарний імунітет, що опосередковує феномен тривалої латентності вірусу. Послаблення інтрагангліонарного імунітету, що відзначається при первинних імунодефицитах або вторинній імуносупресії, є причиною реактивації VZV із латентного стану в сателітних клітинах нервового вузла з поширенням на весь вузол і подальшим ураженням сенсорних нейронів. М. Reichelt зі співавт. показали, що VZV спочатку реактивується із сателітних клітин вузла з подальшим формуванням багатоядерних синцитіальних комплексів, утворених за рахунок злиття гліальних клітин і нейронів, що дозволяє вірусу проникнути в нервові клітини і здійснити міграцію до шкіри вздовж відцентрових волокон периферичних нервів [60]. Тому під час реактивації VZV уражається більшість нейронів ганглія, що пояснює поширений дерматомерний висип на відміну від обмеженої екзантеми HSV1/HSV2-етіології, оскільки ці віруси зазвичай реактивуються в одному або кількох суміжних нейронах і не передаються до інших нервових клітин ганглія, оскільки майже не залучають сателітні клітини [70].
/100-2.jpg)
Натомість у доцентрових нервових волокнах, що прямують від чутливих нервових гангліїв до центральної нервової системи (ЦНС), існує інтранейрональний природжений імунітет у вигляді каскадної системи Toll-like 3 (TLR-3), що розпізнає двоспіральну вірусну РНК й індукує при цьому синтез доімунних інтерферонів під час міграції реактивованого вірусу вздовж нервового волокна у напрямку ЦНС (рис. 3). Ця віростатична система необхідна для профілактики уражень мозку під час реактивації VZV із латентного стану в периферичних нервових гангліях. Пацієнти з природженим дефіцитом TLR-3 або функціонально пов’язаних молекул каскаду є групою ризику щодо розвитку тяжких форм нейроінфекцій, зумовлених альфа-герпесвірусами [83].
/101-1.jpg)
Якщо порушення інтрагангліонарного клітинного імунітету зумовлює збільшену частоту епізодів реактивації вірусу з його відцентровим поширенням й ураженням шкіри й слизових оболонок у вигляді оперізуючого герпесу, то недостатність інтранейронального TLR-3-опосередкованого природженого імунітету створює сприятливі умови для розвитку VZV-індукованих менінгітів, енцефалітів, мієлітів й церебральних васкулопатій унаслідок полегшеної доцентрової міграції збудника.
Власне церебральні васкулопатії розвиваються під час реактивації VZV в гасеровому вузлі трійчастого нерва внаслідок недостатності механізмів інтрагангліонарного і/або інтранейронального захисту. До їх розвитку призводить поширення вірусу вздовж інтракраніальних гілок трійчастого нерва, що іннервують мозкові судини передньої й середньої черепних ямок [66]. При VZV-васкулопатіях спочатку уражаються перицити, що є зовнішнім шаром стінки артерії, звідки згодом вірус проникає до клітин медії та інтими, де розвивається продуктивне запалення з вираженою лімфоцитарною інфільтрацією [79]. D. Jones зі співавт. в нещодавньому експериментальному дослідженні переконливо продемонстрували здатність VZV індукувати прозапальну активність інфікованих фібробластів адвентиції ураженої церебральної судини з продукцією прозапальних цитокінів ІЛ-8 та ІЛ-6 і подальшим поширенням запальної реакції на медію (рис. 4) [34].
/102-1.jpg)
Унаслідок западення товщина медії прогресивно збільшується вогнищево або концентрично залежно від форми васкулопатії, що призводить спочатку до розтягнення, а потім — до надривів внутрішньої еластичної мембрани й ендотелію, які вистилають внутрішню поверхню судини. У місцях таких надривів формуються тромби, що призводять до оклюзії просвіту судини й формування ішемічних лакунарних інфарктів у паренхімі мозку в зоні кровопостачання уражених судин. Такий процес відбувається переважно при запаленні стінок дрібних церебральних артерій. При залученні великих судин нерідко відбувається розшарування запаленої медії з формуванням аневризм або дисекцій артеріальної стінки, що можуть зазнавати подальшого розриву під впливом механічних або гемодинамічних чинників з формуванням геморагічного інсульту, субарахноїдального крововиливу або кровотечі із судин шиї. На відміну від інших інфекційних та автоімунних васкулітів, ендотелій при VZV-васкулопатіях безпосередньо майже не уражається, а запалення в стінці артерії розвивається в напрямку не із середини назовні, а ззовні в середину. Ця особливість зумовлює відсутність ознак системного запалення при VZV-васкулопатіях.
Генетичний дефіцит білка S або продукція автоантитіл до цього протеїну, що зумовлює стан гіперкоагуляції крові, розглядається як чинник предиспозиції до формування тяжких ішемічних уражень мозку під час VZV-васкулопатій [33]. За результатами останнього метааналізу рандомізованих контрольованих клінічних досліджень, проведеного Y. Zhang зі співавт., оперізуючий герпес є незалежним чинником високого ризику розвитку ішемічного інсульту або транзиторної ішемічної атаки (відносний ризик (RR) 1,30, 95% довір–чий інтервал (CI) 1,17–1,46) та інфаркту міокарда в людей (RR = 1,18, 95% CI 1,07–1,30) [84]. Результати нещодавнього великого популяційного клінічного дослідження вказують на різке підвищення ризику розвитку ішемічної хвороби кінцівок внаслідок уражень периферичних артерій після епізоду оперізуючого герпесу (рис. 5) [42].
VZV-васкулопатії можуть формуватися як після вітряної віспи й оперізуючого герпесу, так і поза зв’язком із ними. Слід мати на увазі, що оперізуючий герпес може мати атиповий перебіг у вигляді zoster sine herpete, що зустрічається в 10–20 % випадків [71], хоча, за даними обсерваційного дослідження M. Koskiniemi зі спів–авт., що охоплювало дані 174 пацієнтів із неврологічними ускладненнями при вітряній віспі й оперізуючому герпесі, zoster sine herpete відзначався щонайменше в 44 % випадків. Донедавна не вщухали дискусії щодо відстрочення уражень судин після епізодів вітряної віспи й оперізуючого герпесу [39]. Як продемонстрували нещодавно, VZV здатен персистувати в лейкоцитах крові [72], тобто має властивість, характерну для бета- і гамма-герпесвірусів. Ця особливість пояснює тривалі латентні періоди між епізодом вітряної віспи або оперізуючого герпесу й подальшим неврологічним ураженням, включаючи VZV-васкулопатії, що можуть сягати 6 місяців і більше [70].
Патоморфологія
M.A. Nagel зі співавт. описали патоморфологічні ознаки дифузної васкулопатії дрібних церебральних судин VZV-етіології. Вони зазначили, що характерними проявами були розриви внутрішньої еластичної мембрани, гіперплазія інтими за рахунок клітин, що експресують актин типу α і важкі ланцюги міозину гладеньких м’язів, однак не ендотеліальних клітин, що містять CD31, а також звуження шару гладеньких міоцитів у середній оболонці. На початкових етапах вірус містився в адвентиції, а згодом переміщувався в середню оболонку й інтиму. Це свідчило про реактивацію VZV із ганглія й подальшу трансневральну міграцію до зовнішньої оболонки судин як механізм формування васкулопатії (рис. 6) [53]. У нещодавньому дослідженні M.A. Nagel зі співавт. показали наявність нейтрофілів у адвентиції уражених судин на початковій стадії VZV-васкулопатії, що супроводжувалося потовщенням інтими на ознаками запалення у зоні vaso vasorum. На пізній стадії визначалися антигени VZV у клітинах медії без ознак нейтрофільної інфільтрації. Напевно, вірус використовує імунопривілейовану медію для захисту від імунної відповіді. Т-лімфоцити, макрофаги й меншою мірою — В-клітини спостерігалися в адвентиції та інтимі як на ранній, так і на пізній стадії VZV-васкулопатії. У контрольних зразках, отриманих із здорових судин, патологічних змін виявлено не було [52].
/103-1.jpg)
Класифікація
Ураження судин є унікальною формою VZV-інфекції. Уперше васкулопатію, викликану VZV, описали Cravioto і Feigin в 1959 році, хоча авторам була не відома етіологічна роль даного вірусу при цій патології. Існує більше повідомлень про VZV-васкулопатію мозкових артерій, ніж судин іншої локалізації. На сьогодні відомо, що VZV здатен уражати як дрібні, так середні й великі судини мозку. Частіше за все вірус розмножується одночасно в дрібних і великих церебральних судинах, рідше — тільки в дрібних артеріях, і ще рідше — тільки у великих [53]. Басейн середніх мозкових артерій уражається найчастіше з-поміж інших церебральних судинних басейнів у зв’язку з переважною реактивацією вірусу із гасерового вузла трійчастого нерва [23].
Розрізняють такі клінічні форми VZV-васкулопатії: дифузна багатовогнищева васкулопатія дрібних церебральних артерій, одновогнищевий гранулематозний ангіїт великих артерій, артеріїт скроневої артерії, васкулопатія судин, що живлять корінці краніальних нервів, васкулопатія судин очних яблук, спінальні васкулопатії й васкулопатії периферичних артерій. Нещодавно описана нова форма VZV-васкулопатії — вірус-індукований аортоартеріїт, що нагадує хворобу Такаясу [76]. Про генералізовану форму VZV-васкулопатії говорять при ураженні кількох судинних басейнів у різних компартментах людського організму.
Якщо дифузна васкулопатія дрібних церебральних артерій здебільшого розвивається в імуно-скомпрометованих пацієнтів, то одновогнищевий гранулематозний ангіїт — переважно в імунокомпетентних осіб. Гранулематозний ангіїт частіше реєструють у дітей після епізоду вітряної віспи, тоді як дифузну васкулопатію дрібних церебральних артерій — у дорослих після оперізуючого герпесу або поза зв’язком з ним.
Різні форми ураження судин можуть поєднуватися в одного хворого. Частіше застосовують саме термін «васкулопатія», а не «васкуліт», оскільки при цій формі інфекції відзначаються мінімальні системні ознаки запалення. Це створює разючий контраст з автоімунними васкулітами церебральних артерій. Однак J. Fleming зі співавт. рекомендують розрізняти випадки VZV-індукованої васкулопатії та VZV-васкуліту залежно від наявності або відсутності ознак системного й локального запалення, засвідчуючи, що деякі випадки ураження церебральних судин вірусної етіології можуть нагадувати автоімунні васкуліти [22]. У нашій практиці трапився випадок поступової трансформації VZV-васкулопатії в змішаний розлад у зв’язку з розвитком системного червоного вовчака з продукцією антинуклеарних автоантитіл [4].
Клінічна картина
Дифузна багатовогнищева васкулопатія дрібних церебральних артерій
Ця форма інфекції часто розвивається в імуно-скомпрометованих хворих. При залученні дрібних церебральних судин відзначаються дифузний голов-ний біль, прогресуючі порушення пам’яті й уваги, розлади поведінки, зміна особистості, епілептичний синдром і фокальні неврологічні знаки. У термінальну стадію відзначаються тетрапарез, атаксія, екстрапірамідні прояви, псевдобульбарний синдром, деменція й порушення функції тазових органів за центральним типом — синдромокомплекс, що нагадує лакунарний статус при тяжкому атеросклерозі церебральних судин.
Однак описані випадки, коли відзначався тільки головний біль, що імітував мігрень [23]. Множинні, невеликі за розміром субкортикальні вогнища в білій речовині великих півкуль у басейні середніх мозкових артерій, що відзначаються на МРТ у таких випадках, є лакунарними інфарктами, пов’язаними з тромбуванням запалених судин мозку. Натомість глибші вогнища виникають у разі додаткового ураження олігодендроцитів з індукцією демієлінізації, що призводить до трансформації VZV-нейроінфекції в мультифокальний лейкоенцефаліт [10].
У дітей дифузна васкулопатія дрібних церебральних судин зазвичай розвивається після епізоду вітряної віспи. Слід враховувати, що вітряна віспа може мати безсимптомний або абортивний перебіг. R. Spiegel зі співавт. описали VZV-васкулопатію дрібних церебральних судин у 14-річної дівчинки через 10 років після перенесеної вітряної віспи і через 4 роки після епізоду оперізуючого герпесу. ДНК VZV була ідентифікована в цереброспінальній рідині (CSF) методом полімеразної ланцюгової реакції (ПЛР) [68]. M. Häusler описав васкулопатію дрібних церебральних судин у дитини з множинними кортикальними й субкортикальними інфарктами. Діагноз було підтверджено на підставі аномального індексу співвідношення антитіл до VZV [30]. E. Chiappini зі співавт. повідомили про васкуліт дрібних судин мозку VZV-етіології у дворічної дитини через 20 місяців після перенесеної вітряної віспи [19]. C.J. Selvakumar зі співавт. описали васкулопатію дрібних церебральних судин у дитини через 3 місяці після перенесеної вітряної віспи. Ангіографія демонструвала ураження гілок як середньої, так і задньої церебральної артерії. У лікворі відзначався мононуклеарний плеоцитоз. Були підвищеними концентрації специфічних IgM та IgG у цереброспінальній рідині [66]. T. Okanishi зі співавт. повідомили про 2 випадки васкулопатії в басейні середньої мозкової артерії в дітей після м’яких форм вітряної віспи, які, на перший погляд, характеризувалися сприятливим прогнозом [56]. S.L. West зі співавт. описали природжену форму VZV-васкулопатії дрібних церебральних судин у дитини з прогресуванням хвороби протягом 2 суміжних років [79]. J.F. Caekebeke зі співавт. доповіли про 2 епізоди VZV-васкулопатії з розвитком епізодів геміпарезів через 3 і 9 місяців після вітряної віспи в 5-річного пацієнта. Діагноз підтвердили на підставі виявлення олігоклональних смуг IgG до VZV у цереброспінальній рідині і даних ангіографії церебральних артерій [15].
У дорослих дифузна васкулопатія дрібних церебральних судин зазвичай розвивається після епізоду оперізуючого герпесу, у тому числі форми інфекції у вигляді zoster sine herpete. O. Outteryck зі співавт. описали VZV-васкулопатію дрібних судин у 57-річного пацієнта з ураженням мигдалеподібних тіл і множинними субкортикальними вогнищами в білій речовині півкуль великого мозку. Церебральна артеріографія не демонструвала патологічних змін. ДНК VZV виявили лише в одному з трьох досліджень цереброспінальної рідини методом ПЛР. У лікворі відзначався невисокий мононуклеарний плеоцитоз із невеликою кількістю еритроцитів [58]. J. Katchanov зі співавт. описали 2 випадки VZV-васкуліту дрібних і середніх мозкових артерій у дорослих. Автори звернули увагу на виявлення еритроцитів у лікворі в таких хворих [36]. S.D. Newsome, A. Nath зі співавт. повідомили про васкуліт церебральних судин VZV-етіології у ВІЛ-інфікованого хворого, який прий-мав глюкокортикоїди для лікування синдрому імунної реконституції після призначення антиретровірусної терапії [54]. M.M. Hovens зі співавт. доповіли про розвиток VZV-васкулопатії в реципієнтів алогенної нирки, які приймали імуносупресанти [32]. A. Kronenberg зі співавт. виявили VZV-васкулопатію у ВІЛ-інфікова-ного пацієнта. Автори показали інформативність вивчення феномену інтратекального синтезу антитіл для верифікації клінічного діагнозу [40]. Y. Miyazaki зі співавт. повідомили про випадок поєднання хронічної VZV-васкулопатії з гострим мієлорадикулогангліоменінгоенцефалітом у 66-річної пацієнтки. Ускладненням була субарахноїдальна кровотеча [49].
E. Kaphan зі співавт. описали васкуліт дрібних і середніх мозкових судин VZV-етіології у 37-річної пацієнтки з множинними лакунарними інфарктами в білій речовині півкуль великого мозку. Ангіографія демонструвала нерівномірність просвіту правої внутрішньої парієтальної артерії й васкулярні дефекти в правій парієтальній і лівій окципітальній ділянках. Покращання стану відбулося після комбінованого лікування ацикловіром і преднізолоном [35]. A.N. Russman зі співавт. описали васкулопатію дрібних церебральних судин VZV-етіології з множинними інфарктами мозку в пацієнта зі склеродермією. Діагноз було виставлено на підставі аномального індексу антитіл у лікворі й сироватці крові. Типових уражень шкіри не відзначалося [64].
R. Alvarez зі співавт. описали 3 випадки VZV-васкуло-патії дрібних церебральних судин: два — після herpes zoster ophthalmicus і один — після дисемінованого шкірного герпесу [9]. S. Morgello зі співавт. описали васкуліт лептоменінгеальних артерій VZV-етіології в пацієнта зі СНІДом, який ускладнився мультифокальним лейкоенцефалітом. Тільця Коудрі типу А були ідентифіковані навколо вогнищ демієлінізації в білій речовині півкуль. Імуногістохімічне дослідження показувало наявність антигенів VZV у зонах уражень. Крім того, були виявлені вірусні частинки при електронній мікроскопії [51].
Одновогнищевий гранулематозний ангіїт
Ця форма васкулопатії обумовлена запаленням одного сегмента певної великої артерії з формуванням гранульоми, що обмежує просвіт судини й супроводжується формуванням вторинних тромботичних нашарувань. У клініці має місце картина гострого ішемічного інсульту, який важко відрізнити від атеротромботичного інфаркту мозку, особливо в осіб третьої вікової групи. Здебільшого відзначається пригнічення свідомості різної глибини, геміпарез, геміанестезія, моторна або сенсомоторна афазія.
На МРТ відзначається поодиноке велике гіперінтенсивне вогнище в Т2-зваженому режимі, переважно в ділянці базальних гангліїв і внутрішньої капсули. Ця форма інфекції часто розвивається в імунокомпетентних осіб, нерідко — у дітей невдовзі після епізоду вітряної віспи.
У дітей одновогнищевий гранулематозний ангіїт розвивається через кілька тижнів або місяців після епізоду вітряної віспи. У цілому діагностика в цієї категорії пацієнтів краща, ніж у дорослих осіб, особливо в пацієнтів третьої вікової групи, у яких прояви VZV-васкулопатії плутають із симптомами атеросклерозу судин мозку. N. Danchaivijitr зі співавт. повідомили про гостру внутрішньочерепну кровотечу в дитини після перенесеної вітряної віспи, що було пов’язано з розривом запаленої мозкової судини [20]. M. Kawatani зі співавт. описали одновогнищевий ангіїт VZV-етіо-логії в дитини через 1 місяць після перенесеної вітряної віспи з формуванням інфаркту в ділянці базальних гангліїв з картиною лівобічного геміпарезу й експресивної афазії. Згодом у неї сформувалася мішкоподібна аневризма в передній сполучній артерії. Відзначалося поступове зростання вмісту специфічних IgG у цереброспінальній рідині (метод парних сироваток) [37]. G. Losurdo зі співавт. описали ще 4 випадки одновогнищевого ангіїту VZV-етіології в дітей після вітряної віспи. Відзначалися осередки стенозу або оклюзії в сегменті середньої мозкової артерії з формуванням вогнища ішемічного інсульту в ділянці базальних гангліїв або внутрішньої капсули з картиною геміпарезу й центрального паралічу м’язів обличчя [44]. M. Ueno зі спів–авт. повідомили про ангіїт середньої мозкової артерії в 4-річної пацієнтки через 6 місяців після перенесеного епізоду вітряної віспи. Ангіографічна картина нагадувала хворобу мойя-мойя. Відзначалося аномальне підвищення вмісту специфічних IgG у лікворі [73].
У дорослих однобічний гранулематозний ангіїт формується здебільшого після епізоду оперізуючого герпесу, який може набувати форми zoster sine herpete.
J. Mareedu зі співавт. описали однобічний гранулематозний ангіїт VZV-етіології в пацієнта з ішемічний інфарктом у ділянці базальних гангліїв. У клініці відзначалися лівобічний геміпарез, центральний параліч лицьового нерва й дизартрія. КТ-ангіографія показала оклюзію правої передньої церебральної артерії з конічною дилатацією на її початку, а також виражений стеноз із подальшою фузиформною дилатацією правої середньої мозкової артерії. Лікування в/в ацикловіром привело до усунення ознак васкуліту. Як зазначають автори, летальність у випадку однобічного ангіїту VZV-етіології без лікування становить 25 % випадків [45]. D. Eidelberg зі співавт. повідомили про 3 випадки ангіїту великої церебральної артерії з вторинним тромбозом і формуванням інфаркту мозку. У двох померлих пацієнтів VZV був ідентифікований у середній оболонці судини імунопероксидазним методом. Запалення в зоні перебування VZV було мінімальним [21]. N.M. Ahmad, S.E. Boruchoff описали васкулопатію крупних церебральних судин у 52-річного імунокомпетентного пацієнта з білатеральними великими вогнищами ішемічних інсультів у гемісферах мозку. Ментальний статус хворого покращився тільки після призначення ацикловіру [8].
Інші форми VZV-васкулопатій
Васкулопатія спінальних артерій. Васкулопатію спінальних артерій слід відрізняти від епізодів мієліту VZV-етіології. P.A. McKelvie зі співавт. описали оперізуючий герпес у 67-річної пацієнтки з неходжкінською лімфомою. Згодом відзначався розвиток симптомів ураження спинного мозку з картиною синдрому Броун-Секара. У подальшому мали місце залучення головного мозку й смерть хворої. При автопсії визначено некроз дрібних судин м’якої мозкової оболонки з множинними вогнищами в головному й спинному мозку. У стінках судин мали місце ознаки запалення, відзначалися поодинокі багато–-ядерні клітини Тцанка з інтрануклеарними включеннями Коудрі типу А. Автори зазначали, що до того моменту існувало лише 9 повідомлень про розвиток некротичного васкуліту лептоменінгеальних судин VZV-етіології в людей, хоча наразі накопичено більше даних із цієї проблеми [47].
Васкулопатія скроневої артерії. Васкулопатії скроневих артерій VZV-етіології можуть нагадувати автоімунний гігантоклітинний артеріїт і бути причиною ішемічної невропатії зорового нерва. R. Salazar зі співавт. описали VZV-індуковане запалення скроневої артерії, що призвело до розвитку ішемічної невропатії зорового нерва. Антиген VZV був ідентифікований в адвентиції зазначеної судини, а також уздовж усієї медії [65].
Васкулопатія артерій, що живлять корінці краніальних нервів. VZV може бути причиною розвитку васкулопатій артерій, що живлять корінці черепно-мозкових нервів. При цьому відзначається клінічна картина унілатерального множинного мононевриту краніальних нервів ішемічного генезу згідно з басейном кровопостачання ураженої судини. M. Tecellioglu зі співавт. описали випадок розвитку невритів VII, VIII, IX і X нервів після епізоду zoster sine herpete. ДНК VZV була ідентифікована в лікворі методом ПЛР. Крім того, позитивними були дані методу парних сироваток [71].
Васкулопатія судин очних яблук. Дуже рідко формується ізольоване ураження задньої циліарної артерії або васкулопатія хоріоїдальних судин очей. У таких випадках відзначаються патоморфологічні ознаки гранулематозного ангіїту. Васкуліт ретинальних артерій VZV-етіології описаний у пацієнтів після спленектомії [81]. Також існує повідомлення про ретинальний перифлебіт як наслідок zoster sine herpete [55].
Васкулопатія периферичних артерій. Васкулопатії периферичних артерій VZV-етіології слід відрізняти від автоімунних васкулітів, проявів атеросклерозу й облітеруючого ендартеріїту. I. Uhoda зі співавт. повідомили про лімфоцитарний васкуліт VZV-етіології з ураженням периферичних артерій у пацієнта, який проходив цито–статичну хіміотерапію з приводу лімфоми. Вірус був ідентифікований в ендотеліальних клітинах і дендроцитах шкіри [74]. W.J. Cheatham зі співавт. продемонстрували вірус-індуковане ураження ендотелію периферичних судин різних органів без залучення мозкових артерій. В ендотеліоцитах уражених VZV вісцеральних артерій виявлялися характерні тільця вірусних включень (рис. 7). Пацієнт помер від поліорганної недостатності [17].
/106-1.jpg)
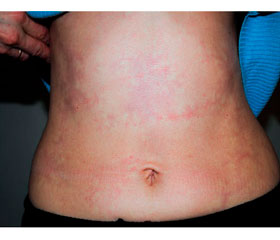
Нами також описаний випадок васкуліту периферичних артерій, викликаного VZV, у молодої пацієнтки з вибірковим дефіцитом природних кілерів. На шкірі уражених передньої поверхні живота (рис. 8А) і нижніх кінцівок (рис. 8Б) відзначалася картина ретикулярного ліведо [6].
J. Massano зі співавт. описали множинні тромбози периферичних артерій у дорослого чоловіка після епізоду вітряної віспи. Особливо тяжкі оклюзії й стенози розташовувалися в басейні стегнової й клубової артерій [46].
/106-2.jpg)
Комбіновані форми нейроінфекції. VZV-нейро-інфекція може рецидивувати кілька разів, трансформуючись в інші клінічні форми, що часом ускладнює діагностику. A. Haug зі співавт. описали 4 рецидиви протягом 11 місяців поспіль у 45-річної жінки. Спочатку розвинувся менінгоенцефаліт з ураженням великих півкуль, потім — васкулопатія дрібних церебральних судин, згодом — –мієліт, і зрештою — стовбуровий енцефаліт [29]. Ми описали трансформацію VZV-нейроінфекції в пацієнта з ізольованим дефіцитом гамма-інтерферону. Спочатку відзначався фокальний грудний мієліт, що анатомічно відповідав дерматомеру, у якому напередодні мав місце оперізуючий герпес. Потім розвинулася васкулопатія дрібних церебральних судин з множинними субкортикальними інфарктами білої речовини півкуль великого мозку, а потім — неврит зорового нерва. Клініка нагадувала розсіяний склероз, тільки зі зворотною динамікою патологічного процесу [4].
Діагностика
Для постановки клінічного діагнозу VZV-васкулопатії необхідно здійснити: а) виявлення реактивованого вірусу в ЦНС; б) нейровізуалізацію для встановлення форми й тяжкості хвороби; в) оцінку імунного статусу для з’ясування причини реактивації опортуністичного вірусу із латентного стану й підбору імунотерапії.
Верифікація діагнозу нейроінфекції. Ідентифікація вірусу в ЦНС можлива на підставі результатів: а) ПЛР ліквору; б) вивчення індексу співвідношення сироваткових і лікворних специфічних IgG; в) вимірювання вмісту специфічних IgМ до VZV у лікворі; г) вивчення олігоклональних смуг імуноглобулінів до антигенів вірусу. Особливістю діагностики VZV-васкулопатій є низька інформативність ПЛР ліквору, результати якого дозволяють виявити вірус щонайбільше в 30 % випадків, оскільки вірус часто не надходить до цереброспінальної рідини. У той же час ідентифікація феномену інтратекального синтезу антитіл до вірусу на підставі аномального індексу співвідношення специфічних IgG у крові й лікворі є інформативною в 60–70 % випадків [24].
M.A. Nagel зі співавт. показали, що визначення аномально підвищеної концентрації специфічних IgG у лікворі інформативніше, ніж ПЛР спинномозкової рідини, у пацієнтів із васкулопатією дрібних церебральних судин [52]. K. Roubalová зі співавт. проаналізували дані 438 пацієнтів з неврологічними ураженнями, які могли бути викликані герпесвірусною інфекцією. Діагноз VZV-нейроінфекції було виставлено на підставі феномену інтратекального синтезу специфічних антитіл у 19,6 % випадків, підвищення вмісту специфічних IgM у сироватці крові — у 17,3 % випадків, ПЛР ліквору — у 9,4 % випадків [63].
Аномальне співвідношення противірусних IgG у сироватці крові й цереброспінальній рідині отримало назву індексу Delpech-Lichtblau [59]. Цей метод може бути використаний у випадку негативних результатів ПЛР, особливо в разі VZV-васкулопатій церебральних артерій. Для цього використовують спеціальні розрахункові формули. Одна з них запропонована P.E. Klapper зі співавт. [38]:
Позитивним результатом вважається значення індексу понад 1,9. Замість альбуміну в даній формулі можна використовувати вміст загального IgG. Припустиме застосування спрощеної розрахункової формули, яку рекомендує експерт Israel Steiner [70]:
У такому разі позитивним результатом вважається значення менше ніж 20 : 1. Якщо рівень індексу менше від одиниці, то результат розглядається як безумовно позитивний, оскільки в такому разі концентрація антитіл у лікворі вища, ніж у сироватці крові, що не може бути пояснено дифузією імуноглобулінів через пошкоджений гематоенцефалічний бар’єр.
Ми удосконалили методику вивчення феномену інтратекальної продукції антитіл, запропонувавши зручнішу для клінічної практики розрахункову формулу [3]:
Позитивним вважається результат, що перевищує 1,9. Така формула досить зручна, коли не відомо, який саме герпесвірус викликав церебральне ураження в пацієнта. Визначаються антитіла принаймні до чотирьох різних герпесвірусів. Запорукою інформативності є наявність хоча б одного негативного результату.
Виявлення олігоклональних смуг імуноглобулінів у лікворі методом ізоелектричного фокусування може бути корисним в окремих випадках VZV-васкулопа-тій, про що є відповідні повідомлення в науковій літературі [66]. M.P. Burgoon зі співавт. показали, що олігоклональні імуноглобуліни при VZV-нейроінфекції специфічні саме до антигенів VZV на відміну від аналогічних смуг при розсіяному склерозі, специфічних до антигенів мієліну. Цей метод може бути вельми інформативним також при підгострому склерозуючому паненцефаліті (рис. 9) [14].
/108-1.jpg)
Встановлення форми й тяжкості васкулопатії. Для нейровізуалізації може бути корисна магнітно-резонансна томографія (МРТ) головного мозку в конвенційних режимах (Т1, Т2, FLAIR). При дифузній васкулопатії дрібних церебральних артерій МРТ головного мозку в конвенційних режимах демонструє множинні гіперінтенсивні в Т2-зваженому режимі невеликі за розміром вогнища в корі й субкортикальних ділянках білої речовини півкуль великого мозку, здебільшого в зоні кровопостачання середньої мозкової артерії, що за природою є лакунарними інфарктами (рис. 10А) [5]. При прогресуванні хвороби з трансформацією в мультифокальний лейкоенцефаліт VZV-етіології деякі з вогнищ розширюються зі втратою чіткості контурів за рахунок приєднання демієлінізації внаслідок проникнення вірусу в паренхіму мозку (рис. 10Б) [23]. Важкими для інтерпретації можуть бути МРТ-знімки мозку при поєднаній патології, наприклад при комбінації ознак VZV-васкулопатії з дисциркуляторною енцефалопатією або хворобою Бісвангера в осіб третьої вікової групи (рис. 10В) [4].
/109-1.jpg)
При одновогнищевому гранулематозному ангіїті VZV-етіології дані МРТ головного мозку в конвенційних режимах відповідають картині гострого ішемічного інсульту атеротромботичного генезу в церебральній гемісфері. Відзначаються гіпоінтенсивні в Т1- та гіпер-інтенсивні в Т-зважених МРТ-режимах поодинокі вогнища середнього або великого розміру в зоні підкіркових ядер, зовнішньої й внутрішніх капсул, таламуса, інсулярної кори, що за природою є ішемічними інфарктами паренхіми головного мозку (рис. 11) [76].
Мультипланарна триміліметрова МРТ головного мозку в Т1-зваженому режимі з контрастним підсиленням демонструє спочатку сегментарне гранулематозне потовщення, а потім аневризматичне розширення й стоншення стінок церебральних артерій із накопиченням контрасту, здебільшого середньої мозкової артерії, у пацієнтів з VZV-васкулопатією у вигляді одновогнищевого гранулематозного ангіїту (рис. 12) [18].
Церебральна ангіографія та МРТ-ангіографія мозкових артерій демонструють сегментарне звуження судини, яке поєднується з постстенотичним розширенням у випадку одновогнищевого гранулематозного ангіїту VZV-етіології [52] та феномен збіднення периферичної судинної сітки за рахунок ампутації гілок дрібних церебральних артерій у разі дифузної багатовогнищевої VZV-васкулопатії церебральних судин [23]. Іноді може знадобитися панангіографія для виявлення екстрацеребральних аневризм, зумовлених генералізованими формами VZV-васкулопатій.
При залученні великих судин голови й шиї при VZV-васкулопатіях ультразвукове допплерографічне дослідження демонструє потовщення комплексу інтима-медіа стінки артерій в зонах ураження, що може бути корисним у діагностиці (рис. 13) [4].
Дослідження ліквору. При дослідженні церебро–спінальної рідини має місце помірний лімфоцитарний плеоцитоз, нормальний або трохи знижений рівень глюкози, незначно підвищений вміст білка [78]. Цей феномен отримав назву клітинно-білкової дисоціації. Однак плеоцитоз відсутній у щонайменше третини пацієнтів із VZV-васкулопатією церебральних судин [23]. Чим глибший клітинний імунодефіцит, тим менша ймовірність виявлення лімфоцитарного плеоцитозу при VZV-нейроінфекції.
Оцінка імунного статусу. Оцінка імунного статусу є важливою для з’ясування причини реактивації вірусу з латентного стану й підбору імунотерапії. Нерідко можна виявити дефіцит природних кілерів, природних кілерних Т-лімфоцитів і/або CD8+ цитотоксичних Т-клітин, що зумовлює критичне послаблення інтрагангліонарного контролю над VZV [6, 12]. Нещодавно продемонстровано, що генетичний дефіцит фолатного циклу є поширеною причиною первинного дефіциту кілерних лімфоцитів в організмі людини [2]. Пацієнти з VZV-васкулопатіями мають проходити дослідження на ВІЛ.
Патоморфологічні дослідження. Гігантські багатоядерні клітини Тцанка можна виявити в лімфоцитарних депозитах навколо лептоменінгеальних судин, а також у VZV-індукованих зонах некрозу білої речовини мозку [26]. Виявляються також інтрануклеарні тільця Коудрі типу А [62]. VZV ідентифікують в ураженій тканині мозку або стінках церебральних судин імуногістохімічним методом або за допомогою гібридизації in situ [25], а також з використанням електронної мікроскопії [51]. D.I. Victor, W.R. Green рекомендують робити біопсію темпоральної артерії для ідентифікації вірус-індукованого васкуліту, що є причиною відстроченого геміпарезу при офтальмічному оперізуючому герпесі [77]. Дані патоморфологічого аналізу уражених стінок церебральних судин при VZV-васкулопатіях наведені на рис. 14 [52].
Диференціальна діагностика
У молодих пацієнтів може знадобитися проведення диференціальної діагностики VZV-васкулопатії із розсіяним склерозом, а в осіб третього віку — з дисциркуляторною енцефалопатією атеросклеротичного генезу.
Відсутність перивентрикулярних, субпіальних і суб–епендимальних вогнищ при дифузній багатовогнищевій VZV-васкулопатії дрібних церебральних артерій допомагає в диференціальній діагностиці з розсіяним склерозом. У разі нейроінфекції вогнища лакунарних інфарктів, на відміну від перивентрикулярно локалізованих осередків демієлінізації при розсіяному склерозі, тяжіють до кори, розташовуючись переважно субкортикально, рідше — інтракортикально. Крім того, вогнища в білій речовині півкуль при VZV-васкулопатії не накопичують контраст, за винятком прогредієнтних форм із трансформацією в мультифокальний лейкоенцефаліт [51].
При лакунарному статусі атеротромботичного генезу ішемічні осередки інфарктів локалізуються в ділянці базальних гангліїв і внутрішньої капсули, зонах суміжного кровопостачання судинних басейнів або ділянках кровопостачання артерій, уражених оклюзійним атеросклерозом. Важливими для диференціальної діагностики є візуалізація церебральних судин, ідентифікація й оцінка розміру атеросклеротичних бляшок, встановлення гемодинамічної значимості виявлених атеросклеротичних уражень, а також зіставлення локалізації інфарктів і зон судинної оклюзії [4].
Важкою може бути диференціальна діагностика вірусної васкулопатії й васкуліту церебральнх судин при системному червоному вовчаку. При автоімунній хворобі зазвичай є позитивними маркери системного запалення й специфічні серологічні тести, наприклад антинуклеарні автоантитіла й автоантитіла до двоспіральної ДНК. S. Osiro, N. Salomon доповіли про поєднання в одного пацієнта VZV-васкулопатії й системного червоного вовчака (рис. 15) [57].
J. Gutierrez, G. Ortiz провели диференціальну діагностику між ВІЛ-васкулопатією й васкулітом церебральних судин VZV-етіології. Як зазначають автори, у випадку ВІЛ-васкулопатії формуються поодинокі великі за площею центровані вогнища ішемії в півкулях великого мозку, тоді як при дифузному багатовогнищевому VZV-васкуліті мають місце множинні дрібні осередки лакунарних інфарктів переважно в субкортикальних ділянках білої речовини гемісфер, здебільшого в зоні кровопостачання середньої церебральної артерії [28].
У випадку легкої дифузної VZV-васкулопатії дрібних церебральних судин може сформуватися поодиноке вогнище лакунарного інфаркту, подібне до проявів одновогнищевого гранулематозного анігіїту VZV-етіології, як це описали J. Venhovens зі співавт. [75], тому часом проведення диференціальної діагностики навіть між різними формами VZV-васкулопатій є досить складним завданням.
Ускладнення
Типовим ускладненням одновогнищевого ангіїту великих церебральних артерій VZV-етіології є аневризми й дисекції каротидних артерій, що можуть ставати причиною геморагічних уражень (рис. 16) [41]. При дифузній васкулопатії дрібних артерій мозку неодноразово описані випадки тромбозу венозних синусів мозку (рис. 17) [33]. Відомі також випадки розвитку автоімунних демієлінізуючих уражень у вигляді розсіяного склерозу або розсіяного енцефаломієліту після епізоду VZV-нейроінфекції. Такі ускладнення слід відрізняти від випадків прогресування VZV-васкулопатії до багатовогнищевого лейкоенцефаліту VZV-етіології [51].
Лікування
Запорукою запобігання розвитку VZV-васку-лопатій є правильне й своєчасне лікування вітряної віспи й оперізуючого герпесу. Важливою є ідентифікація й компенсація імунодефіцитних хвороб, що дозволяє покращити імунний нагляд за ендогенним вірусом.
Доказова база противірусних хіміопрепаратів з групи ациклічних аналогів гуанозину, насамперед ацикловіру як препарату першої лінії терапії, у комбінації з глюкокортикостероїдами при VZV-васкулопатіях базується на результатах невеликих клінічних досліджень і повідомлень про клінічні випадки (рівень доказовості С). Хоча в цілому при альфа-герпесвірусних інфекціях доказовість призначення ацикловіру вища й сягає рівня В.
D. Gilden зі співавт. рекомендують застосування ацикловіру в/в в дозі 10–15 мг/кг 3 рази на добу протягом 14 діб поряд із преднізолоном у дозі 1 мг/кг протягом 5 діб при VZV-васкулопатії в людей. Після цього доцільною є пероральна терапія валацикловіром у дозі 3 г/добу протягом 1–2 місяців під клініко-інструментальним контролем [23]. Згідно з повідомленням R. Spiegel зі співавт., терапія за допомогою ацикловіру й високої дози метилпреднізолону призвела до часткового позитивного ефекту при тяжкій, загрозливій для життя VZV-васкулопатії дрібних церебральних судин у дитини [68]. C. Yasuda зі співавт. з успіхом використали терапію ацикловіром в/в в дозі 750 мг/день протягом 14 діб із подальшим місячним курсом валацикловіру в дозі 3 мг/добу per os при VZV-васкулопатії, що проявлялася у вигляді ішемічного інфаркту мозку в зоні правої внутрішньої капсули та інтракраніальної аневризми, у молодого пацієнта зі СНІДом, кількість CD4+ T-лімфоцитів у крові якого була нижчою від 200/μl [82].
У випадку резистентності до ацикловіру можуть бути застосовані ганцикловір, фоскарнет або цидофовір як противірусні хіміопрепарати другої лінії терапії. У пацієнтів з тяжкою імуносупресією слід уникати призначення глюкокортикостероїдів у високій дозі, оскільки ті можуть поглибити імунодефіцит [23].
Результати щонайменше 5 подвійних сліпих плацебо-контрольованих рандомізованих клінічних досліджень вказують на ефективність і безпечність препаратів природних, лімфобластоїдних і рекомбінантних альфа2а- і альфа-2b-інтерферонів людини в дозі від 1 до 5 млн МО в/м або підшкірно через день № 10–30 при реактивованих альфа-герпесвірусних інфекціях [11, 43, 48, 50, 80]. Ці дані, а також сучасні уявлення щодо ключової ролі альфа-інтерферонів у реалізації механізмів інтрагангліонарного й інтранейронального імунітету проти латентного VZV можуть бути використані для обґрунтування призначення препаратів альфа-інтерферонів у складі комплексної терапії VZV-васкулопатій у людей. Доказова база для призначення альфа-інтерферонів зростає при виявленні дефіциту природних кілерів, природних кілерних Т-лімфоцитів і/або цитотоксичних CD8+ Т-клітин у пацієнта з VZV-васкулопатією, оскільки ці імунотерапевтичні препарати продемонстрували ефективність при зазначених імунних дисфункціях у невеликих контрольованих дослідженнях і повідомленнях про клінічні випадки [6]. Натомість у разі гуморальних імунодефіцитів, включаючи загальний варіабельний імунодефіцит, може знадобитися додаткове призначення 5% або 10% препаратів нормального в/в імуноглобуліну людини в місячній дозі від 200 до 800 мг/кг маси тіла для заміщення дефіциту ендогенних імуноглобулінів.
Пацієнти зі СНІДом ВІЛ-етіології потребують високоактивної антиретровірусної терапії, а особи з медикамент-індукованою вторинною імуносупресією — заходів з раціоналізації імуносупресивних лікувальних втручань.
У разі коморбідного дефіциту білка S необхідно застосовувати в гострий період VZV-васкулопатії гепарин, а потім продовжувати проводити профілактику тромбоутворення непрямими антикоагулянтами [33].
Профілактика
Своєчасне виявлення й лікування первинних і вторинних імунодефіцитів може забезпечити запобігання випадкам тяжких форм VZV-нейроінфекцій в імуноскомпрометованих осіб, що має бути основою неспецифічної профілактики VZV-васкулопатій у людей.
Розроблені живі атенуйовані вакцини проти VZV, що можуть бути використані для планової активної імунізації людей. Упровадження програми загальнонаціональної активної імунізації проти VZV у США дозволило різко скоротити кількість неврологічних ускладнень, спричинених цим убіквітарним вірусом, у тому числі VZV-васкулопатій церебральних артерій.
Пасивна імунізація при VZV-інфекції проводиться препаратами 10% нормального імуноглобуліну для в/м введення, який містить достатню кількість антитіл до VZV із крові здорових донорів, і специфічними гіперімунними імуноглобулінами проти VZV для в/м і в/в введень. Centers for Disease Control and Prevention повідомляють, що з 2011 року дозволено використовувати нормальний або специфічний анти-VZV-імуноглобулін людини пацієнтам з групи високого ризику протягом перших 10 діб після ймовірного інфікування для екстреної постконтактної пасивної профілактики VZV-інфекції [16].
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Евтушенко С.К. Герпесвирусные церебральные васкулиты в генезе нарушений мозгового кровообращения у детей // Неврология раннего детского возраста. — К., 2016. — С. 158-184.
2. Мальцев Д.В. Оценка иммунного статуса у детей с расстройствами аутистического спектра, ассоциированными с генетическим дефицитом фолатного цикла // Клінічна імунологія, алергологія, інфектологія. — 2016. — № 5(94). — С. 23-28.
3. Мальцев Д.В., Казмірчук В.Є. Модифікована методика порівняльних серологічних досліджень для діагностики персистуючої герпесвірусної нейроінфекції // Лабораторна діагностика. — 2011. — № 3(57). — С. 24-30.
4. Мальцев Д.В., Климчук В.В. Труднощі у діагностиці неврологічних ускладнень VZV-інфекції: випадки з клінічної практики // Український медичний часопис. — 2011. — 5(85). — С. 122-127.
5. Мальцев Д.В., Казмірчук В.Є., Євтушенко С.К. До питання сучасної клініко-вірусологічної класифікації герпесвірусних нейроінфекцій // Міжнародний неврологічний журнал. — 2012. — № 2(48). — С. 14-28.
6. Мальцев Д.В., Недопако Я.Я. Дефіцит природних кілерів: гетерогенність, клініка, діагностика, лікування, клінічні приклади // Український медичний часопис. — 2013. — № 2(94). — С. 129-142.
7. Adler H., Beland J.L., Del-Pan N.C., Kobzik L. et al. In the absence of T cells, natural killer cells protect from mortality due to HSV-1 encephalitis // J. Neuroimmunol. — 1999. — Vol. 93(1–2). — P. 208-213.
8. Ahmad N.M., Boruchoff S.E. Multiple cerebral infarcts due to varicella-zoster virus large-vessel vasculopathy in an immunocompetent adult without skin involvement // Clin. Infect. Dis. — 2003. — Vol. 37(1). — e16-18.
9. Alvarez R., Graus F., Abós J. et al. Postherpetic vasculopathy. A study of 3 cases in immunosuppressed patients // Med. Clin. (Barc.). — 1990. — Vol. 95(20). — P. 782-724.
10. Amlie-Lefond C., Kleinschmidt-DeMasters B.K., Mahalingam R. et al. The vasculopathy of varicella-zoster virus encephalitis // Ann. Neurol. — 1995. — Vol. 37(6). — P. 784-790.
11. Arvin A.M., Kushner J.H., Feldman S. et al. Human leukocyte interferon for the treatment of varicella in children with cancer // N. Eng. J. Med. — 1982. — Vol. 306(13). — P. 761-765.
12. Backström E., Chambers B.J., Kristensson K., Ljunggren H.G. Direct NK cell-mediated lysis of syngenic dorsal root ganglia neurons in vitro // J. Immunol. — 2000. — Vol. 165(9). — P. 4895-4900.
13. Bastian F.O., Rabson A.S., Yee C.L., Tralka T.S. Herpesvirus hominis: isolation from human trigeminal ganglion // Science. — 1972. — Vol. 178(4058). — P. 306-307.
14. Burgoon M.P., Hammack B.N., Owens G.P. et al. Oligoclonal immunoglobulins in cerebrospinal fluid during varicella zoster virus (VZV) vasculopathy are directed against VZV // Ann. Neurol. — 2003. — Vol. 54(4). — P. 459-463.
15. Caekebeke J.F., Peters A.C., Vandvik B. et al. Cerebral vasculopathy associated with primary varicella infection // Arch. Neurol. — 1990. — Vol. 47(9). — P. 1033-1035.
16. Centers for Disease Control and Prevention. Closure of vari-cella-zoster virus-containing vaccines pregnancy registry — United States, 2013 // MMWR Morb. Mortal. Wkly Rep. — 2014. — Vol. 63(33). — P. 732-733.
17. Cheatham W.J., Dolan T.F. Jr, Dower J.C., Weller T.H. Varicella: report of two fatal cases with necropsy, virus isolation, and serologic studies // Am. J. Pathol. — 1956. — Vol. 32(5). — P. 1015-1035.
18. Cheng-Ching E., Jones S., Hui F.K. et al. High-resolution MRI vessel wall imaging in varicella zoster virus vasculopathy // J. Neurol. Sci. — 2015. — Vol. 351(1–2). — P. 168-173.
19. Chiappini E., Calabri G., Galli L. et al. Varicella-zoster virus acquired at 4 months of age reactivates at 24 months and causes encephalitis // J. Pediatr. — 2002. — Vol. 140(2). — P. 250-251.
20. Danchaivijitr N., Miravet E., Saunders D.E. et al. Post-vari-cella intracranial haemorrhage in a child // Dev. Med. Child. Neurol. — 2006. — Vol. 48(2). — P. 139-142.
21. Eidelberg D., Sotrel A., Horoupian D.S. et al. Thrombotic cerebral vasculopathy associated with herpes zoster // Ann. Neurol. — 1986. — Vol. 19(1). — P. 7-14.
22. Fleming J., Fogo A., Haider S. Varicella zoster virus brachioplexitis associated with granulomatous vasculopathy // Clin. Exp. Dermatol. — 2013. — Vol. 38(4). — P. 378-381.
23. Gilden D., Nagel M.A., Cohrs R.J., Mahalingam R. et al. The variegate neurological manifestations of varicella zoster virus infection // Curr. Neurol. Neurosci Rep. — 2013. — Vol. 13(9). — P. 374.
24. Gilden D.H., Bennett J.L., Kleinschmidt-DeMasters B.K. et al. The value of cerebrospinal fluid antiviral antibody in the diagnosis of neurologic disease produced by varicella zoster virus // J. Neurol. Sci. — 1998. — Vol. 159(2). — P. 140-144.
25. Gray F., Bélec L., Lescs M.C., Chrétien F. et al. Varicella-zoster virus infection of the central nervous system in the acquired immune deficiency syndrome // Brain. — 1994. — Vol. 117 (Pt 5). — P. 987-999.
26. Gray F., Mohr M., Rozenberg F., Belec L. et al. Varicella-zoster virus encephalitis in acquired immunodeficiency syndrome: report of four cases // Neuropathol. Appl. Neurobiol. — 1992. — Vol. 18(5). — P. 502-514.
27. Grubor-Bauk B., Simmons A., Mayrhofer G., Speck P.G. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR // J. Immunol. — 2003. — Vol. 170(3). — P. 1430-1434.
28. Gutierrez J., Ortiz G. HIV/AIDS patients with HIV vasculopathy and VZV vasculitis: a case series // Clin. Neuroradiol. — 2011. — Vol. 21(3). — P. 145-151.
29. Haug A., Mahalingam R., Cohrs R.J. et al. Recurrent polymorphonuclear pleocytosis with increased red blood cells caused by varicella zoster virus infection of the central nervous system: Case report and review of the literature // J. Neurol. Sci. — 2010. — Vol. 292(1–2). — P. 85-88.
30. Häusler M., Schaade L., Kemény S. et al. Encephalitis related to primary varicella-zoster virus infection in immunocompetent children // J. Neurol. Sci. — 2002. — Vol. 195(2). — P. 111-116.
31. Henin D., Gervaz E., Seilhean D. et al. Pathologic anatomy of cytomegalovirus encephalomyelitis and varicella-zona virus encephalomyelitis // J. Neuroradiol. — 1995. — Vol. 22(3). — P. 180-183.
32. Hovens M.M., Vaessen N., Sijpkens Y.W., de Fijter J.W. Unusual presentation of central nervous system manifestations of Varicella zoster virus vasculopathy in renal transplant recipients // Transpl. Infect. Dis. — 2007. — Vol. 9(3). — P. 237-240.
33. Imam S.F., Lodhi O.U.H., Fatima Z. et al. A Unique Case of Acute Cerebral Venous Sinus Thrombosis Secondary to Primary Varicella Zoster Virus Infection // Cureus. — 2017. — Vol. 9(9). — e1693.
34. Jones D., Neff C.P., Palmer B.E. et al. Varicella zoster virus-infected cerebrovascular cells produce a proinflammatory environment // Neurol. Neuroimmunol. Neuroinflamm. — 2017. — Vol. 4(5). — e382.
35. Kaphan E., Witjas T., Feuillet L. et al. Multiple strokes after chickenpox primo-infection in an adult // Rev. Neurol. (Paris). — 2005. — Vol. 161(8–9). — P. 840-843.
36. Katchanov J., Siebert E., Klingebiel R., Endres M. Infectious vasculopathy of intracranial large- and medium-sized vessels in neurological intensive care unit: a clinico-radiological study // Neurocrit. Care. — 2010. — Vol. 12(3). — P. 369-374.
37. Kawatani M., Nakai A., Okuno T. et al. A case of intracranial saccular aneurysm after primary varicella zoster virus infection // Brain Dev. — 2012. — Vol. 34(1). — P. 80-82.
38. Klapper P.E., Bailey A.S., Longson M. et al. Meningo-encephalitis caused by Coxsackievirus group B type 2. Diagnosis confirmed by measuring intrathecal antibody // J. Infect. — 1984. — Vol. 8(3). — P. 227-231.
39. Koskiniemi M., Piiparinen H., Rantalaiho T. et al. Acute central nervous system complications in varicella zoster virus infections // J. Clin. Virol. — 2002. — Vol. 25(3). — P. 293-301.
40. Kronenberg A., Schupbach R., Schuknecht B. et al. Multifocal vasculopathy due to Varicella-Zoster Virus (VZV): serial analysis of VZV DNA and intrathecal synthesis of VZV antibody in cerebrospinal fluid // Clin. Infect. Dis. — 2002. — Vol. 35(3). — P. 330-333.
41. Liberman A.L., Nagel M.A., Hurley M.C. et al. Rapid deve-lopment of 9 cerebral aneurysms in varicella-zoster virus vasculopathy // Neurology. — 2014. — Vol. 82(23). — P. 2139-2141.
42. Lin T.Y., Yang F.C., Lin C.L. et al. Herpes zoster infection increases the risk of peripheral arterial disease: A nationwide cohort study // Medicine (Baltimore). — 2016. — Vol. 95(35). — e4480.
43. Liu Y., Wang J., Li S. Clinical trial of recombinant alpha-2a interferon in the treatment of herpes zoster // Zhongguo Yi Xue Ke Xue Yuan Xue Bao. — 1998. — Vol. 20(4). — P. 264-266.
44. Losurdo G., Bertoluzzo L., Canale F. et al. Varicella and its complications as cause of hospitalization // Infez. Med. — 2005. — Vol. 13(4). — P. 229-234.
45. Mareedu J., Hanumaiah R.G., Hale E., Habte-Gabr E. Varicella zoster vasculopathy // J. Int. Assoc. Physicians AIDS Care (Chic.). — 2011. — Vol. 10(3). — P. 144-145.
46. Massano J., Ferreira D., Toledo T. et al. Stroke and multiple peripheral thrombotic events in an adult with varicella // Eur. J. Neurol. — 2008. — Vol. 15(10). — e90-91.
47. McKelvie P.A., Collins S., Thyagarajan D. et al. Meningoencephalomyelitis with vasculitis due to varicella zoster virus: a case report and review of the literature // Pathology. — 2002. — Vol. 34(1). — P. 88-93.
48. Merigan T.C., Rand K.H, Pollard R.B. et al. Human leukocyte interferon for the treatment of herpes zoster in patients with cancer // N. Engl. J. Med. — 1978. — Vol. 298(18). — P. 981-987.
49. Miyazaki Y., Riku Y., Goto Y.. VZV vasculopathy associated with myelo-radiculoganglio-meningo-encephalitis: an autopsy case of an immunocompetent 66-year-old male // J. Neurol. Sci. — 2008. — Vol. 275(1–2). — P. 42-45.
50. Montero Mora P., Colín D., González Espinosa A., Almeida Arvizu V. Interferon alpha 2b in pain caused by herpes zoster. Preliminary report // Rev. Alerg. Mex. — 1996. — Vol. 43(6). — P. 148-151.
51. Morgello S., Block G.A., Price R.W., Petito C.K. et al. Varicella-zoster virus leukoencephalitis and cerebral vasculopathy // Arch. Pathol. Lab. Med. — 1988. — Vol. 112(2). — P. 173-177.
52. Nagel M.A., Gilden D. Complications of varicella zoster virus reactivation // Curr. Treat. Options Neurol. — 2013. — Vol. 15(4). — P. 439-453.
53. Nagel M.A., Traktinskiy I., Azarkh Y. et al. Varicella zoster virus vasculopathy: analysis of virus-infected arteries // Neurology. — 2011. — Vol. 77(4). — P. 364-370.
54. Newsome S.D., Nath A. Varicella-zoster virus vasculopathy and central nervous system immune reconstitution inflammatory syndrome with human immunodeficiency virus infection treated with steroids // J. Neurovirol. — 2009. — Vol. 15(3). — P. 288-291.
55. Noda Y., Nakazawa M., Takahashi D. et al. Retinal periphlebitis as zoster sine herpete // Arch. Ophthalmol. — 2001. — Vol. 119(10). — P. 1550-1552.
56. Okanishi T., Kondo A., Inoue T. et al. Bilateral middle cerebral artery infarctions following mild varicella infection: a case report // Brain Dev. — 2009. — Vol. 31(1). — P. 86-89.
57. Osiro S., Salomon N. Varicella-zoster virus (VZV) multifocal vasculopathy in a patient with systemic lupus erythematosus — a diagnostic and treatment dilemma // IDCases. — 2017. — Vol. 8. — P. 81-83.
58. Outteryck O., Sénéchal O., Berteloot D. et al. Cerebral vasculitis secondary to Varicella-Zoster virus infection // Rev. Neurol. (Paris). — 2005. — Vol. 161(8–9). — P. 836-839.
59. Parisi G., Squadrone N.P., Rojo S. Herpes simplex virus infection in pregnancy: epidemiologic, diagnostic and therapeutic data. An unusual case of HSV-1 in monozygotic twins // Minerva Pediatr. — 1992. — Vol. 44(10). — P. 459-467.
60. Reichelt M., Zerboni L., Arvin A.M. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia // J. Virol. — 2008. — Vol. 82(8). — P. 3971-3983.
61. Richter E.R., Dias J.K., Gilbert J.E. 2nd, Atherton S.S. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck // J. Infect. Dis. — 2009. — Vol. 200(12). — P. 1901-1906.
62. Rosenblum M.K. Bulbar encephalitis complicating trigeminal zoster in the acquired immune deficiency syndrome // Hum. Pathol. — 1989. — Vol. 20(3). — P. 292-295.
63. Roubalová K., Suchánková A., Bojar M. et al. Detection of active varicella-zoster virus (VZV) infection in patients with neurological complications // Klin. Mikrobiol. Infekc. Lek. — 2007. — Vol. 13(3). — P. 109-114.
64. Russman A.N., Lederman RJ, Calabrese L.H. et al. Multifocal varicella-zoster virus vasculopathy without rash // Arch. Neurol. — 2003. — Vol. 60(11). — P. 1607-1609.
65. Salazar R., Russman A.N., Nagel M.A. et al. Varicella zoster virus ischemic optic neuropathy and subclinical temporal artery involvement // Arch. Neurol. — 2011. — Vol. 68(4). — P. 517-520.
66. Selvakumar C.J., Justin C., Gnaneswaran T.R., Chandrase-karan M. Post varicella angiopathy // J. Assoc. Physicians India. — 2010. — Vol. 58. — P. 572-574.
67. Shiihara H. Neurological complications of varicella-zoster virus (VZV) infection // No To Hattatsu. — 1993. — Vol. 25(2). — P. 128-134.
68. Spiegel R., Miron D., Lumelsky D., Horovitz Y. Severe meningoencephalitis due to late reactivation of Varicella-Zoster virus in an immunocompetent child // J. Child. Neurol. — 2010. — Vol. 25(1). — P. 87-90.
69. Steain M., Sutherland J.P., Rodriguez M. et al. Analysis of T cell responses during active varicella-zoster virus reactivation in human ganglia // J. Virol. — 2014. — Vol. 88(5). — P. 2704-2716.
70. Steiner I., Kennedy P.G., Pachner A.R. The neurotropic herpes viruses: herpes simplex and varicella-zoster // Lancet Neurol. — 2007. — Vol. 6(11). — P. 1015-1028.
71. Tecellioglu M., Kamisli S., Erbay M.F. et al. A Rare Presentation of Cranial Polyneuropathy Without Rash Caused by Varicella Zoster Virus // Med. Arch. — 2017. — Vol. 71(4). — P. 293-295.
72. Terada K., Niizuma T., Kawano S. et al. Detection of varicella-zoster virus DNA in peripheral mononuclear cells from patients with Ramsay Hunt syndrome or zoster sine herpete // J. Med. Virol. — 1998. — Vol. 56(4). — P. 359-363.
73. Ueno M., Oka A., Koeda T. et al. Unilateral occlusion of the middle cerebral artery after varicella-zoster virus infection // Brain Dev. — 2002. — Vol. 24(2). — P. 106-108.
74. Uhoda I., Piérard-Franchimont C., Piérard G.E. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement // Dermatology. — 2000. — Vol. 200(2). — P. 173-175.
75. Venhovens J., Stelten B., Feyen B.F. et al. Ischemic Stroke as a Complication of Varicella Zoster Encephalitis: A Case Report With Detailed EEG Discussion // Clin. EEG Neurosci. — 2014. — Vol. 45(4). — P. 310-314.
76. Verma R., Lalla R., Patil T.B. Extensive extracranial and intracranial Varicella zoster vasculopathy // BMJ Case Rep. — 2012. — Vol. 2012. — pii: bcr2012006845.
77. Victor D.I., Green W.R. Temporal artery biopsy in herpes zoster ophthalmicus with delayed arteritis // Am. J. Ophthalmol. — 1976. — Vol. 82(4). — P. 628-630.
78. Weerkamp N.J., Keizer K., Boel C.H., de Rijk M.C. et al. Meningoencephalitis caused by varicella zoster virus // Ned. Tijdschr. Geneeskd. — 2010. — Vol. 154. — A1575.
79. West S.L., Newton R.W., Baildam E.M. et al. Recurrent hemiplegia associated with cerebral vasculopathy following third trimester maternal herpes zoster infection // Dev. Med. Child Neurol. — 2006. — Vol. 48(12). — P. 991-993.
80. Winston D.J., Eron L.J., Ho M., Pazin G., Kessler H., Pottage J.C., Gallagher J., Sartiano G., Ho W.G., Champlin R.E. Recombinant interferon alpha-2a for treatment of herpes zoster in immunosuppressed patients with cancer // Am. J. Med. — 1988. — Vol. 85(2). — P. 147-151.
81. Wolf A.H., Thurau S.R., Kook D., Haritoglou C. Ocular manifestation of primary VZV infection in a splenectomized patient // Ocul. Immunol. Inflamm. — 2008. — Vol. 16(4). — P. 199-201.
82. Yasuda C., Okada K., Ohnari N. et al. Cerebral infarction and intracranial aneurysm related to the reactivation of varicella zoster virus in a Japanese acquired immunodeficiency syndrome (AIDS) patient // Rinsho Shinkeigaku. — 2013. — Vol. 53(9). — P. 701-705.
83. Zhang S.Y., Jouanguy E., Ugolini S. et al. TLR3 deficiency in patients with herpes simplex encephalitis // Science. — 2007. — Vol. 317(5844). — P. 1522-527.
84. Zhang Y., Luo G., Huang Y. et al. Risk of Stroke/Transient Ischemic Attack or Myocardial Infarction with Herpes Zoster: A Systematic Review and Meta-Analysis // J. Stroke Cerebrovasc. Dis. — 2017. — Vol. 26(8). — P. 1807-1816.

/107-1.jpg)
/107-2.jpg)
/107-3.jpg)
/109-2.jpg)
/110-1.jpg)
/111-1.jpg)
/111-2.jpg)
/112-1.jpg)
/112-2.jpg)