Введение
Современное определение бронхолегочной дисплазии (БЛД) очертило критерии диагностики. БЛД — диффузное паренхиматозное заболевание легких преждевременно рожденных детей с острой дыхательной недостаточностью в неонатальном периоде, которым проводилась искусственная вентиляция легких (ИВЛ) и/или кислородотерапия более 28 дней жизни [6]. Бронхолегочная дисплазия в настоящее время имеет относительно благоприятный прогноз [5]. Вместе с тем приходится констатировать факт развития тяжелого респираторного ди–стресс-синдрома (РДС) в периоде новорожденности у доношенных (редко — преждевременно рожденных детей). РДС обычно обусловливает необходимость длительной механической вентиляции легких и экстракорпоральной мембранной оксигенации (ЭКМО). Однако терапия неэффективна, и в первые месяцы жизни наступает летальный исход. Одной из причин развития данного патологического состояния является интерстициальное заболевание легких (ИЗЛ), ассоциированное с диффузным нарушением их развития [4].
Исследователи Европейского респираторного общества и Американской торакальной ассоциации отмечают низкую регистрацию интерстициальных заболеваний среди детской популяции (0,7–1,32–3,6 случая ИЗЛ на 1 млн обследованных детей), что объясняют несовершенными стандартами диагностики, разнообразной морфологией и неспецифической клиникой ИЗЛ, которая маскируется за проявлениями иной респираторной или даже кардиальной патологии. Так, часть заболеваний из группы ИЗЛ у детей раннего возраста достоверно не диагностируется ни с помощью оценки бронхоальвеолярного лаважа, ни по результатам компьютерной томографии легких. Биопсия и анализ аутопсийного материала легких при условии достаточной квалификации патологоанатома может пролить свет на причину РДС у новорожденного [4].
Целью обзора являлась систематизация мировых представлений об интерстициальных заболеваниях легких (диффузных паренхиматозных заболеваниях легких), связанных с их развитием, на примере клинического наблюдения.
Клиническое наблюдение
Представляем клиническое наблюдение ребенка 3 месяцев, родившегося в срок 38 недель, дыхательная недостаточность которого требовала длительной механической вентиляции с невозможностью перехода на более щадящие режимы кислородной поддержки. Из анамнеза жизни и заболевания известно, что ребенок от IV беременности (первые III беременности — здоровые дети), протекавшей на фоне угрозы срыва, кольпита и маловодия. Роды IV, в срок 38 недель гестации, самостоятельные. Безвод–ный промежуток — 88 часов. Масса тела при рождении — 3080 г. Длина тела — 46 см. По шкале Апгар 1 минута — 6 баллов, 5 минут — 7–8 баллов. Признаки респираторного дистресс-синдрома появились через 32 часа после рождения, в связи с чем начата механическая вентиляция легких (инспираторный поток — 10 л/мин, положительное давление конца выдоха (PEEP) — 9–12 см вод.ст., фракция кислорода во вдыхаемом воздухе (FiO2) — 40–60 %). Мать, 33 года, страдала ожирением II степени, отец, 36 лет, здоров. При осмотре в 3 месяца состояние ребенка было тяжелое, обусловленное дыхательной недостаточностью. Механическая вентиляция легких, инспираторный поток — 12 л/мин, PEEP — 11 см вод.ст., FiO2 — 48 %. Без дотации 48% О2 сатурация 84 %. Температура тела 36,8 °С, частота сердечных сокращений 156 в 1 минуту, артериальное давление 85/45 мм рт.ст. Грудная клетка ригидна, уплощена в переднезаднем направлении, с втяжением нижних отделов при дыхании. Перкуторно над легкими — коробочный звук с притуплением паравертебрально. Аускультативно дыхание ослаблено, выдох удлиненный, выслушивалась крепитация с 2 сторон. Границы относительной сердечной тупости: правая — парастернальная линия, верхняя — II межреберье, левая — на 2 см кнаружи от среднеключичной линии. Тоны сердца ритмичные, приглушенные, акцент 2-го тона над легочной артерией. Живот умеренно вздут. Печень +2,5–3,0 см ниже края реберной дуги.
По данным компьютерной томографии органов грудной клетки высокого разрешения обнаружено «матовое стекло» над всей поверхностью легких, гиперпневматоз 17,5.
Ребенку проведена допплер-эхокардиография (Д-ЭхоКГ). Выявлена значительная дилатация правых камер и асинергия сокращений миокарда задней стенки левого желудочка. Открытое овальное окно 3 мм mean PAP — 57 мм рт.ст. Диастолическая дисфункция левого желудочка.
В ходе мониторинга проводилась дифференциальная диагностика между диффузными нарушениями развития паренхимы, аномалией строения легких, гиперплазией нейроэндокринных клеток, пульмональным интерстициальным гликогенозом, мутацией протеинов сурфактанта. Установлен диагноз «интерстициальное заболевание легких детей до 2 лет: диффузное нарушение развития паренхимы легких».
Динамика состояния. На 11, 17, 26, 39, 45, 59, 77, 89, 96-е сутки жизни у ребенка, находившегося на ИВЛ, отмечались внезапные эпизоды ухудшения состояния в виде нарастания дыхательной недостаточности, появление разлитого цианоза кожи и слизистых оболочек, снижение насыщения гемоглобина кислородом до 50 %. Данные эпизоды требовали ужесточения параметров ИВЛ, повышения концентрации кислорода до 50–100 %. Через 1–1,5 часа от начала данных эпизодов состояние больного улучшалось, исчезал цианоз, повышалась сатурация. На 98-е сутки отмечалось два эпизода внезапного ухудшения состояния, первый из которых сопровождался брадикардией до 50 ударов в 1 минуту и снижением сатурации кислорода до 10 %, во время второго наряду со всеми вышеперечисленными симптомами возникло резкое снижение артериального давления. После комплекса реанимационных мероприятий в полном объеме был зафиксирован летальный исход.
Диагноз, установленный в результате аутопсии: «интерстициальное заболевание легких детей до 2 лет; диффузное нарушение развития паренхимы легких (альвеолокапиллярная дисплазия)».
Обсуждение
Терминология. Интерстициальные заболевания легких — гетерогенная группа респираторных заболеваний, основой которых является нарушение респираторной функции легких. Однако в настоящее время ученые всего мира все больше склоняются к определению «диффузные паренхиматозные заболевания легких», так как в данную группу входит патология, ассоциированная как с поражением интерстиция, так и альвеолярного эпителия, пульмонального капиллярного эндотелия и терминальных бронхиол.
Классификация диффузных паренхиматозных заболеваний легких, характерных для детей ≤ 2 лет, включает диффузные нарушения развития, аномалии строения легких, специфические состояния неопределенной или недостаточно определенной этиологии, мутации протеинов сурфактанта и сопутствующие расстройства (табл. 1).
Прогностически неблагоприятной группой диффузных паренхиматозных заболеваний считаются диффузные нарушения развития легких. Как представлено выше, к диффузным нарушениям развития легких принято относить ацинарную дисплазию, врожденную альвеолярную дисплазию, альвеолокапиллярную дисплазию с недостаточностью легочных вен.
История открытия диффузных нарушений развития легких. Макмахон в 1947 году впервые описал врожденную альвеолокапиллярную дисплазию легких, а в 1981 году Дженни и его коллеги представили данные о развитии тяжелого респираторного дистресс-синдрома у ребенка, который родился доношенным. Новорожденный умер в возрасте 40 часов жизни. Патологоанатомическое исследование показало торможение роста альвеол, вен и альвеолярных капилляров. Так, с конца ХХ века критерии диффузных нарушений развития легких начали более распространенно использоваться в практике неонатологов и педиатров, а на аутопсии обращали внимание на характерные патоморфологические изменения. В некоторых наблюдениях продолжительность жизни продлевалась до семи месяцев [7].
Эпидемиология. Частота диффузных нарушений развития легких еще не известна, но есть вероятность потенциально большей распространенности заболевания, чем ранее ожидалось. Кроме того, представляется вероятным, что некоторые случаи, первоначально классифицированные как идиопатическая персистирующая легочная гипертензия новорожденного (persistent pulmonary hypertension of the newborn, PPHN), фактически были диффузными нарушениями развития легких.
Определенно известно, что группа диффузных паренхиматозных заболеваний легких, связанных с их развитием, распространена во всем мире, а частота регистрации в регионах обусловлена знаниями неонатологов и патологоанатомов клиники и патоморфологии мальформации. Имеется тенденция к преобладанию мужской популяции (60 %). Более 90 % детей с диффузными нарушениями развития легких рождаются в срок, из них у 60 % цианоз и респираторная недостаточность возникают в течение первых 48 часов после рождения.
Врожденная ацинарная дисплазия — самая тяжелая форма мальформации легочной паренхимы, которая отличается появлением дыхательной недостаточности вскоре после рождения, прогредиентным течением и летальным исходом в течение первых дней жизни [1]. Сообщается о мутации гена TBX4 в генезе ацинарной дисплазии [3].
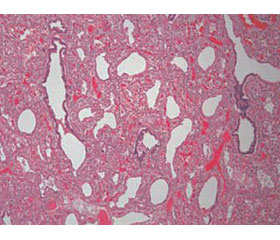
Патоморфология. Гистологически паренхима легких характеризуется задержкой развития легких на поздней каналикулярной или ранней саккулярной стадиях (схематическое изображение представлено на рис. 1). Развитие дыхательных путей ограничено бронхиальными и бронхиолярными структурами. Описательный термин «псевдогландулярный» используется в связи с особенностью строения эпителия респираторного тракта. Он простой столбчатый, с субъядерными включениями гликогена, схожий с железистой тканью. Ацинус ребенка содержит 3–7 (обычно 14–16) респираторных бронхиол, ацинарные протоки и альвеолярные саккулы (мешочки) [1, 2].
/185-1.jpg)
/186-1.jpg)
Диагностические критерии. Ацинарная дисплазия может выявляться как у доношенных, так и преждевременно рожденных детей. Первым клиническим признаком является тяжелый респираторный дистресс-синдром, который возникает вскоре после рождения. Часто имеет место высокая легочная гипертензия. Течение заболевания тяжелое, вентиляционная поддержка обычно неэффективна. Новорожденные умирают в первые дни жизни.
Дополнительные критерии. Характерными рентгенологическими признаками (чаще выявляются по данным компьютерной томографии легких высокого разрешения) является гипоплазия с признаками отека легких. Клинические проявления респираторного дистресс-синдрома обычно не разрешаются, формируется картина «матового стекла».
Биопсия демонстрирует повышенную плотность легочной ткани с признаками гипоплазии. Кисты не визуализируются. Характерна дисплазия ацинуса дистальнее респираторных бронхиол, обусловленная полным отсутствием мешочков и альвеол. В редких случаях аномально развитые саккулы и альвеолы присутствуют в небольшом количестве.
Врожденная альвеолярная дисплазия. Отличительной особенностью альвеолярной дисплазии считается удовлетворительное формирование дистальной легочной паренхимы и недоразвитие мезенхимы, что реализуется в большом расстоянии между капилляром и альвеолой. То есть создается впечатление, что мальформация начинается с дистальных отделов бронхиол в структурах, примыкающих к плевре и междольковым перегородкам [1]. Лобулярная архитектура легких не выражена, воздушные пространства более многочисленны, чем при ацинарной дисплазии. Саккулы выложены кубическим эпителием, а субъядерный гликоген исчезает. Появляются вторичные мезенхимальные гребни, содержащие капилляры.
Патоморфология. Терминальные и респираторные бронхиолы развиты удовлетворительно, междольковые бронхиолы сопровождаются легочными артериями и венами, а альвеолы отличаются тонкостенностью, однокамерностью и простыми контурами. Мальформация паренхимы представлена диффузно по всей поверхности легких. Часто диагностируется гипоплазия обоих легких.
Диагностические критерии. Так же, как и при ацинарной дисплазии, первым клиническим признаком является тяжелый респираторный дистресс-синдром новорожденных (РДСН). Часто РДСН сопровождается легочной артериальной гипертензией. Течение заболевания прогредиентное, механическая вентиляция легких более эффективна, чем при ацинарной дисплазии. При обеспечении вентиляционной поддержки и ЭКМО продолжительность жизни может достигать нескольких недель, редко — месяцев.
Дополнительные критерии. Так же, как и при ацинарной дисплазии, характерным рентгенологическим признаком является гипоплазия с признаками отека легких. В последующем в легочной паренхиме формируются «матовое стекло» и диффузный пневмофиброз.
Биопсия. Развитие дистальной легочной паренхимы удовлетворительное. Недоразвитие мезенхимы реализуется в недоразвитии альвеол и большом расстоянии между капилляром и альвеолой.
Альвеолярная симплификация — отличительная черта аномалий альвеоляризации (alveolar simplification, AGAs). Основной морфологической особенностью данной мальформации является расширение и упрощение альвеолярных пространств, которые становятся округлыми или удлиненными и не имеют перегородки. В некоторых случаях могут быть кистозные изменения с интерстициальным фиброзом. Сами альвеолярные пространства обычно пусты, а пневмоциты типа II отсутствуют. Существуют два общих вторичных изменения, которые могут сопровождать альвеолярную симплификацию: легочный интерстициальный гликогеноз (pulmonary interstitial glycogenosis, PIG), альвеолокапиллярная дисплазия (alveolar capillary dysplasia, ACD).
Вместе с тем AGAs может формироваться у детей с неонатальной пневмонией или сепсисом, диабетической фетопатией. В последнее время AGAs описана у детей с дефицитом TTF-1 (синдром, включающий сочетание церебрального дисгенеза, тиреоидной дисфункции, диффузное нарушение развития легких). Альвеолярная симплификация выявляется при гипоплазии легких, врожденных пороках сердца, хромосомных аномалиях (особенно трисомии по 21-й хромосоме). Отметим, что для синдрома Дауна характерно субплевральное альвеолярное «упрощение» [9].
Врожденная альвеолокапиллярная дисплазия с дисфункцией пульмональных вен (alveolar capillary dysplasia with misalignment of the pulmonary veins, ACD/MPV) — заболевание, обусловленное абнормальной васкуляризацией альвеол, торможением роста альвеол, вен и альвеолярных капилляров [7]. То есть поражены наиболее дистальные отделы легочной паренхимы, что обусловлено мальформацией мезенхимы.
В мире зарегистрировано более 100 случаев ACD/MPV. Большинство из них спорадические, но в 10 % случаев заболевание наследуется. Доказано влияние FOXF1 гена на хромосоме 16q24.1 (z 40%). ACD/MPV может развиваться у новорожденных с трисомией по 21-й хромосоме. Часто диагноз альвеолярно-капиллярной дисплазии скрывается за идиопатической легочной гипертензией новорожденных. В отличие от идиопатической легочной гипертензии новорожденных ACD/MPV имеет прогредиентное течение, летальный исход происходит в первые месяцы жизни, а механическая вентиляция и ЭКМО неэффективны [7].
Патоморфология. Причиной ACD/MPV считают недоразвитость и небольшое число легочных капилляров, недостаточный их контакт с альвеолярным эпителием и утолщение мышечного слоя артериол. Нередко диагностируется наличие у ребенка других пороков развития. Возможен фенотип с более высокой плотностью капилляров и меньшим расстоянием к альвеолярному эпителию. Распространенность этого более мягкого фенотипа пока неизвестна [7].
Диагностические критерии. С первых суток жизни у новорожденного обычно появляются и нарастают тахипноэ и цианоз как клинические проявления респираторного дистресс-синдрома, сопровождающиеся легочной артериальной гипертензией. В отличие от бронхолегочной дисплазии клиническая симптоматика нарастает постепенно и имеет необратимое фатальное течение. Течение заболевания с тяжелой гипоксемией и метаболическим ацидозом. Без трансплантации легких обычно наступает летальный исход, обусловленный правожелудочковой и дыхательной недостаточностью. Курсы пульмональных вазодилататоров, механической вентиляции, экстракорпоральной жизненной поддержки неэффективны [8].
У 50–80 % детей возможны пороки развития гастроинтестинальной, мочевыделительной и сердечно-сосудистой систем.
Дополнительные критерии. По данным компьютерной томографии высокого разрешения характерно раннее появление «матового стекла» и диффузного пневмофиброза. Данная симптоматика в представленном клиническом наблюдении появилась на четвертой неделе жизни и нарастала в течение жизни ребенка. Часто выявляется «обеднение» легочного рисунка по периферии и высокий артериобронхиальный коэффициент (соотношение диаметра артерии и бронха ≥ 1,5) [1, 7].
Катетеризация правых отделов сердца — золотой стандарт при определении давления в легочной артерии. Для ACD/MPV характерно отсутствие капиллярной перфузии. Но катетеризация сердца является инвазивной манипуляцией и не была проведена ребенку в связи с высоким риском интраоперационной летальности.
Трансторакальная допплер-эхокардиография — обязательный метод диагностики легочной гипертензии за счет достаточной точности определения систолического, диастолического и среднего давления в легочной артерии. Основным параметром определения легочной гипертензии по Д-ЭхоКГ является систолическое давление в легочной артерии (pulmonary arterial systolic pressure, systolic-PAP), определяющееся путем регистрации пиковой скорости трикуспидальной регургитации (velocity tricuspid regurgitation, Vtr). В представленном клиническом наблюдении наравне с тяжелой дыхательной недостаточностью выявлена высокая systolic-PAP (57 мм рт.ст.), что и позволило заподозрить диффузное нарушение развития легких [1, 7].
Биопсия. Окончательный диагноз основан на гистологическом исследовании легочной ткани патологоанатомом с опытом работы в диагностике паренхиматозных мальформаций легких. Однако только в 10 % случаев диагноз был установлен с помощью биопсии [1, 7]. Важно отметить возможность имитации симплификации альвеол при воздействии формалина.
Лечение диффузных нарушений развития легких
Единственным эффективным способом лечения ИЗЛ, связанным с их развитием, считается пересадка легких. В мире в год проводится около 2 пересадок легких из 200 возможных у детей с диффузными нарушениями развития. Несколько продлить период ожидания может экстракорпоральная мембранная оксигенация. Использование специфической терапии легочной артериальной гипертензии: оксида азота, простаноидов, ингибиторов эндотелиновых рецепторов и фосфодиэстеразы малоэффективно [1, 2].
Таким образом, имея сходную клиническую симптоматику с бронхолегочной дисплазией, идиопатической легочной гипертензией новорожденных и другими интерстициальными заболеваниями у детей до 2 лет, ацинарная, альвеолярная и альвеолокапиллярная дисплазия отличаются ранним появлением респираторного дистресс-синдрома, высокой легочной гипертензией и характерными патоморфологическими изменениями. К сожалению, в настоящее время прижизненная диагностика данной группы заболеваний еще ограниченна, а диффузные нарушения развития легких являются редкой и пока фатальной патологией для новорожденных. Вместе с тем развитие таргетной терапии и трансплантологии может открыть новые перспективы в продлении жизни маленьких пациентов.
Конфликт интересов. Автор заявляет об отсутствии какого-либо конфликта интересов при подготовке данной статьи.

/184-1.jpg)