Введение
Ph-негативные миелопролиферативные неоплазмы (МПН) трансформируются в острую миелобластную лейкемию (ОМЛ) или миелодиспластический синдром (МДС) в 5–10 % случаев обычно через 10 и более лет от момента диагностики заболевания [1–4]. Бластная прогрессия Ph-негативных МПН является мультифакторной, но может быть вызвана вовлечением аберрантных метилированных генов-супрессоров опухолевого роста [5], включая отрицательные регуляторы (SOCS-1 и SOCS-3) JAK-сигнальной трансдукции и активации латентных факторов транскрипции (STAT), находящихся в цитоплазме в неактивном состоянии [6–8]. Эти трансформации характеризуются рефрактерностью к химиотерапии (ХТ), высоким риском рецидивов, следствием чего является плохая выживаемость (обычно 3–4 месяца) [9, 10]. При МПН механизмы, лежащие в основе прогрессии в МДС или ОМЛ, недостаточно известны. Установлено, что одним из факторов эволюции миелоидной метаплазии является гиперметилирование генов p15INK4, p16INK4a семейства INK4 [6].
Аберрантные CpG-островки гиперметилирования, возникающие в процессе регуляции генов-супрессоров опухолевого роста, активируют их, играя значительную роль в патогенезе различных опухолей человека [11]. Так, в продвинутой фазе сублейкемического миелоза (ИМФ) обнаружены метилирование генов p15INK4B, p16INK4A и рецептора ретиноевой кислоты β [6, 12].
После открытия в 1960 году специфической хромосомной транслокации t(9; 22)(q34; q11) и идентификации химерного гена BCR/ABL при хроническом миелоидном лейкозе усилия многих ученых были направлены на поиск различных мутаций при других формах МПН с целью разработки новых препаратов для лечения этих заболеваний. Эти надежды были реализованы в 2005 году, когда четыре группы исследователей сообщили о наличии единственной мутации в тирозинкиназе JAK2 (JAK2V617F) у пациентов с истинной полицитемией (ИП), эссенциальной тромбоцитемией (ЭТ) и ИМФ [13–16]. Мутация JAK2V617F при классических хронических миелопролиферативных заболеваниях появляется вследствие замены нуклеотида гуанина на тимин в 1849-м нуклеотиде 14-го экзона. В результате этого синтезируется аминокислота фенилаланин вместо валина в положении 617 JH2 области JAK2-киназы.
В 2005 г. на конференции Американского общества гематологов ведущие специалисты по МПН, обсудив эти новые данные, обосновали роль мутации JAK2V617F в патогенезе ИП, ЭТ и ИМФ. В последующем эта мутация была выявлена и при других миелоидных заболеваниях, включая атипические МПЗ И МДС [17, 18], а также в клеточных линиях острых лейкемий, включая эритролейкемию.
Открытие мутации JAK2V617F — главное крупное достижение в понимании молекулярного патогенеза классических МПН, однако до настоящего времени остаются ограниченными исследования прогностической значимости JAK2V617F. Проведены два исследования с включением 150 пациентов с ЭТ и 117 пациентов с ИМФ. Анализ полученных данных не выявил прогностической значимости JAK2V617F-мутации и продолжительности заболевания [19]. Снижение выживаемости отдельные авторы связали с повышенным риском лейкемической трансформации у пациентов с ИМФ, позитивных по JAK2V617F-мутации [20]. Однако предположительная связь между наличием мутации и бластной или фибротической трансформацией при МПН не была подтверждена другими исследованиями [19].
Открытие JAK2V617F-мутации при МПН инициировало разработку фармакологических ингибиторов JAK2 [20]. Так, US Food and Drug Administration (US FDA) для терапии ИМФ рекомендовали Ruxolitinib как первый класс JAK-ингибиторов [21, 22].
Обнадеживающие результаты были получены при исследовании других таргетных препаратов (SAR302503 и LY2784544), а также эпигенетических модификаторов — панобиностата (Panobinostatinum, ингибитор пандиацетилазы), действие которых направлено против костномозгового фиброза [23–25]. Применение этих препаратов показано в основном в хронической фазе заболевания, тогда как лечение фазы акселерации или бластного криза (БК) остается до настоящего времени практически неразработанным.
Изучение активных эпигенетических изменений, открытие ингибиторов ДНК-метилтрансферазы, которая индуцирует реактивацию метилированных генов, способствовали разработке препаратов, действие которых направлено на гипометилирование: 5-azacitidine (azacitidine; Vidaza, Pharmion, Boulder, CO, USA) and 5-aza-2′-deoxycitidine (decitabine; Dacogen, MGI Pharma Inc., Bloomington, MN, USA). Оба эти агента были вначале введены в практику FDA (США) для лечения пациентов с МДС [26, 27].
Результаты контролируемых рандомизированных исследований показали высокую эффективность гипометилирующих препаратов у пациентов с МДС высокого риска [26, 28]. При исследовании эффективности 5-азацитидина у пациентов с МПН при первичном ИМФ или ИМФ на фоне прогрессирования ЭТ и ИП доказано наличие глобального гипометилирования, однако значительного клинического улучшения не получено [28]. Тем не менее в этих работах отсутствовали данные о применении 5-азацитидина для лечения ОМЛ и МДС, которые развились при трансформации МПН.
Результаты первых сообщений по применению 5-азацитидина у пациентов с Ph-негативными МПН при прогрессии в МДС/ОМЛ, которые получили лечение гипометилирующим агентом [29–32], показали эффективность гипометилирующих препаратов при трансформации хронических МПН в МДС/ОМЛ. Так, ответ на азацитидин был зарегистрирован у 64 % пациентов с прогрессией в МДС и у 38 % пациентов — при прогрессии в ОМЛ. Число ответов было значительно выше у пациентов с ЭТ, чем у пациентов с ИП. Ответ ассоциировался с возвратом хронической фазы МПН. Число ответов на 5-азацитидин при МДС после прогрессии МПН и идиопатическом МДС оказалось сходным [27, 29]. Среднее количество циклов 5-азацитидина равнялось 6 (колебания от 1 до 28). 35 пациентов (65 %) получали 5-азацитидин в дозе 75 мг/м2/день в течение 7 дней каждые 28 дней. 19 пациентов (35 %) получали редуцированную дозу препарата (100 мг/день в течение 7 дней или 75 мг/м2/день в течение 5 дней). Редуцирование дозы препарата до 100 мг в сутки проводили у лиц пожилого возраста, а также при наличии тяжелой сопутствующей патологии. Полная ремиссия (ПР) развилась у 13 человек (24 %), ПР с цитопенией — у одного пациента (2 %), минимальная ПР — у 3 пациентов (6 %), частичная ремиссия — у 6 (11 %) и стабилизация заболевания — у 5 человек (9 %).
Рецидив после проведенного лечения 5-азацитидином у 23 из 28 человек зарегистрирован в среднем после 6 месяцев (1,5–22 месяца) и у 6 пациентов — после 15 месяцев (10–34 месяца).
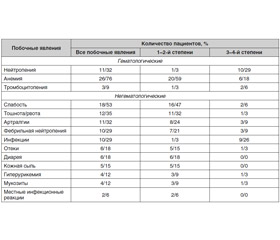
У 34 пациентов, включенных в исследование [30], общее количество циклов 5-азацитидина составило 142. Только у 14 (41 %) пациентов наблюдалось 18 эпизодов токсичности 3–4-й степени. Наиболее часто встречались анемия, слабость, тошнота, артралгия, нейтропения и фебрильная нейтропения (табл. 1). У 16 (47 %) пациентов возникла необходимость снизить дозу 5-азацитидина: 14 (41 %) пациентам — до 50 мг/м2/день и 2 (6 %) — до 25 мг/м2/день. Один пациент умер от сердечной недостаточности, у другого, с тромбоцитопенией 4-й степени, перед получением 5-азацитидина развилась субдуральная гематома в течение первого курса терапии, которая была успешно вылечена. У 33 (97 %) пациентов терапия азацитидином была прервана вследствие отсутствия ответа (n = 10), развития ремиссии (n = 7), токсичности 3–4-й степени (n = 6), трансформации в ОМЛ (n = 2), аллогенной трансплантации костного мозга (ТКМ) (n = 1), смерти (n = 1), решения пациента (n = 1) и интеркуррентной медицинской проблемы (n = 5).
/24-1.jpg)
Таким образом, первые исследования по использованию гипометилирующих препаратов свидетельствуют о терапевтическом эффекте 5-азацитидина у пациентов с трансформацией МПН в ОМЛ/МДС.
Учитывая эти данные, мы приняли решение об использовании 5-азацитидина (Виндуза, компания «Др. Реддис Лабораториз») для лечения бластной трансформации у пациента с ИМФ.
Клинический случай
Пациент С., 1974 года рождения, считает себя больным с 27 июля 2010 года, когда во время управления автомобилем почувствовал резкую жгучую боль за грудиной. По скорой помощи поступил в кардиологическое отделение 3-й больницы г. Кременчуга. В приемном покое у пациента наступила остановка сердца. Проводились реанимационные мероприятия. Кроме того, введены тромболитические препараты (тенектеплаза 10 000 ЕД). При УЗИ сердца обнаружены снижение фракции выброса (ФВ) левого желудочка (ЛЖ) до 39 % и аневризма межжелудочковой перегородки.
15.11.2010 поступил в киевскую городскую больницу «Киевский городской центр сердца», где был выставлен диагноз: ишемическая болезнь сердца (ИБС), постинфарктный (Q-ИМ 27.07.10) и атеросклеротический кардиосклероз. Аневризма верхушечной области ЛЖ с тромбом. Сердечная недостаточность (СН) IIА со сниженной систолической функцией ЛЖ. ФК 3. В анализе крови был обнаружен лейкоцитоз (12,2 • 109/л), показатели количественного содержания эритроцитов (4,84 • 1012/л) и уровня гемоглобина (Hb) (135 г/л) оставались в пределах нормальных величин.
19.11.10 произведена операция: МКШ-1 (передняя межжелудочковая ветвь левой коронарной артерии), аневризмэктомия ЛЖ, тромбэктомия из ЛЖ. После–операционный период протекал без особенностей.
В течение 2 лет наблюдался у кардиолога. В анализах крови постоянно отмечался лейкоцитоз (колебания от 9,0 до 13,0 • 109/л), беспокоило чувство дискомфорта в брюшной полости. В начале 2012 года пациент начал отмечать появление зуда кожи после теплой ванны, а в феврале 2012 года при исследовании периферической крови обнаружен выраженный тромбоцитоз. С этого же времени было обнаружено увеличение размеров селезенки.
В августе 2012 года в гематологическом отделении Главного военного клинического госпиталя (ГВКГ) была выполнена трепанобиопсия, и по данным гистологического исследования костного мозга (КМ) выставлен диагноз: идиопатический (первичный) миелофиброз.
С октября 2012 года по май 2013 года получал терапию рИФН в дозе 3 млн МЕ 3 раза в неделю. С начала марта 2013 года в связи с прогрессированием заболевания назначена гидроксимочевина в дозе 1,0 г ежедневно. Через месяц после начала циторедуктивной терапии пациент начал предъявлять жалобы на резкую слабость. При осмотре обнаружена желтушность кожных покровов, иктеричность склер. При исследовании анализа крови выявлена нормоцитарная, нормохромная анемия тяжелой степени (эритроциты (эр.) — 2,34 • 1012/л, Hb — 72 г/л), лейкоциты (л.) — 4,4 • 109/л, палочкоядерные нейтрофилы (п/я) — 2 %, сегментоядерные нейтрофилы (с/я) — 50 %, лимфоциты (лимф.) — 37 %, моноциты (м.) — 9 %, тромбоциты (тромб.) — 229,0, ретикулоциты — 7 %. Проба Кумбса (прямая и непрямая) была положительной (2+). Уровень билирубина составил 36 мкмоль/л. На основании клинико-лабораторных данных выставлен диагноз аутоиммунной гемолитической анемии (АИГА) тяжелой степени. Был начат курс пульс-терапии метилпреднизолоном с последующим применением метилпреднизолона в малой дозе.
В апреле 2014 года пациент отметил прогрессирующее увеличение размеров селезенки. Была начата терапия рИФН и продолжена терапия гидроксимочевиной и преднизолоном. Лечение получал в течение 1,5 месяца. Однако в июне 2014 года вновь отметил появление общей слабости, повышенной утомляемости, появилась выраженная одышка и сердцебиение при физической нагрузке, отеки на нижних конечностях. При исследовании крови обнаружено прогрессирование АИГА. С 01.07.2014 по 09.07.2014, с 11.08.2014 по 15.08.2014, с 27.10.2014 по 29.10.2014 проходил лечение в клинике гематологии НВМКЦ «ГВКГ» — проводились трансфузии эритроцитной массы на фоне пульс-терапии глюкокортикоидными препаратами.
В 2015 году пациент был направлен на консультацию в ГУ «Национальный институт хирургии и трансплантологии имени А.А. Шалимова» для решения вопроса о возможности проведения спленэктомии. После обследования был выставлен диагноз: идиопатический миелофиброз. Аутоиммунная гемолитическая анемия в стадии нестойкой ремиссии. Спленомегалия. Синдром гиперспленизма. ИБС: постинфарктный (2010) кардиосклероз. Состояние после КШ-1, БЛНПГ. СН IIА ст. со сниженной систолической функцией (ФВ — 36 %). Хронический эзофагит.
29.09.2015 проведена спленэктомия (размеры селезенки 26 × 17 × 8 см). Послеоперационный период протекал без осложнений. Анализ крови при выписке из стационара: л. — 7,2 • 109/л ; эр. — 5,2 • 1012/л; Hb — 136 г/л, тромб. — 356,0 • 109/л. Формула крови (%): п/я — 5, с/я — 56, эозинофилы (э.) — 4, м. — 15, лимф. — 20. Выписан в удовлетворительном состоянии с рекомендациями приема гидроксимочевины по 1000 мг через день, метилпреднизолона по 72 мг/сутки с постепенным уменьшением дозы, омепразола по 1 капсуле 2 раза в день, препаратов калия.
В ноябре 2017 года проходил лечение в кардиологическом отделении по поводу повторного инфаркта миокарда, который был обусловлен тромбированием стента. Получал консервативную терапию.
В январе 2018 года перенес простудное заболевание, принимал антибактериальную терапию. В конце февраля 2018 года пациент отметил появление общей слабости и повышенной утомляемости, появились боли в костях. Был госпитализирован в гематологическое отделение НВМКЦ «ГВКГ». Миелограмма: (28.03.18): бласты — 25 %, миелоциты — 5 %, п/я — 1 %, с/я — 4 %, э. — 1 %, м. — 7 %, лимф. — 40 %, плазматические клетки — 2 %. Клетки эритроидного ряда — 15 %.
Выставлен диагноз: идиопатический миелофиброз, бластный криз, аутоиммунная гемолитическая анемия, рецидивирующее течение. Состояние после спленэктомии (09.2015). ИБС, постинфарктный –(Q-ИМ 27.07.10) и атеросклеротический кардиосклероз. Аневризма верхушечной области ЛЖ с тромбом. СН IIА со сниженной систолической функцией левого желудочка. ФК 3.
Проведен первый курс терапии 5-азацитидином (Виндуза, компания «Др. Реддис Лабораториз»), после которого развилась миелодепрессия и фебрильная нейтропения.
Динамика показателей крови представлена в табл. 2.
После 1-го курса терапии 5-азацитидином (с 04.04 по 10.04.2018 г.) самочувствие пациента улучшилось; бласты в периферической крови отсутствовали, происходило постепенное снижение количества лимфоцитов. 18.04.2018 был выписан из стационара с рекомендацией наблюдения гематологом по месту жительства. Продолжал принимать антибактериальную терапию, однако через 5 дней после выписки из стационара был отмечен подъем температуры тела до 38,5 °С. В период с 26.04.2018 по 14.05.2018 проходил лечение в клинике гематологии НВМКЦ «ГВКГ» по поводу цитостатической миелодепрессии, внегоспитальной пневмонии и острого флеботромбоза правой верхней конечности.
С 22.05 по 28.05.2018 г. проведен 2-й курс 5-азацитидина в дозе 100 мг внутривенно. Динамика показателей периферической крови представлена в табл. 3.
Введение 5-азацитидина перенес без осложнений. На фоне введения Виндузы принимал глюкокортикоидные препараты, ривароксабан по 20 мг в сутки, клопидогрель, предуктал, аллопуринол, пантопразол, торасемид. Выписан в удовлетворительном состоянии с рекомендациями наблюдения кардиологом, гематологом и эндокринологом по месту жительства.
25.06.2018 поступил в клинику гематологии для проведения очередного (третьего) курса терапии 5-азацитидином в удовлетворительном состоянии. По результатам проведенного обследования в данную госпитализацию у пациента диагностирован первичный остеоартроз правого голеностопного сустава. Рент–генстадия II. ПФС II.
Проба Кумбса (26.06.18) прямая — негативная, непрямая — негативная.
Миелограмма (26.06.18): общее количество миелокариоцитов снижено, количество бластных клеток в пределах нормы (1 %). Гранулоцитарный росток в норме, но наблюдается увеличение количества сегментоядерных нейтрофилов. Эритроцитопоэз преимущественно нормоцитарный. В препаратах значительное количество «отшнурованных» молодых тромбоцитов.
В удовлетворительном состоянии пациент был выписан из стационара с рекомендациями продолжить наблюдение гематологом по месту жительства и через 28 дней продолжить терапию 5-азацитидином (4-й курс). Однако пациент отказался от дальнейшего лечения ввиду финансовых трудностей. Мы оценивали ответ на терапию после проведения трех курсов лечения 5-азацитидином. Согласно критериям, разработанным International Working Group for Myelofibrosis Research and Therapy (IWGMRT) [32], у нашего пациента после 3 курсов терапии 5-азацитидином развилась частичная ремиссия.
Обсуждение
Метилирование цитозина является эпигенетическим феноменом, который коррелирует со снижением экспрессии генов-супрессоров опухолевого роста (tumor suppression genes), вследствие чего происходит селективное увеличение количества опухолевых клеток [25]. Известно, что трансформация Ph-негативных МПН в МДС или ОМЛ сочетается с плохим ответом на ХМ и характеризуется короткой продолжительностью жизни больных. S. Thepot и соавторы [31] представили результаты II фазы исследования активности ингибитора ДНК метилтрансферазы 5-азацитидина у 54 пациентов с МПН (табл. 5).
Пациенты получали 5-азацитидин в дозе 75 мг/м2/день подкожно ежедневно в течение 7 дней каждые 4 недели. 35,6 % пациентов имели патологическую цитогенетику, и у 19 % обнаружена мутация JAK2V617F.
Оказалось, что прогностическим фактором для развития ПР был тип МПН. Так, пациенты с ЭТ ответили лучше, чем пациенты с ИП; количество пациентов с ИМФ было небольшое, ответ наблюдался у 4 из 7 человек. У пациентов с МДС при трансформации и у пациентов с ЭТ как проявлением первичной МПН наблюдалось более высокое число ПР, чем у пациентов с ОМЛ при трансформации и ИП как проявлением первичной МПН соответственно.
Согласно результатам предыдущих исследований по применению азацитидина при МДС [27, 28], комплексный кариотип не является неблагоприятным фактором для ответа. При трансформации МПН часто наблюдается комплекс хромосомных аномалий, при этом частота ответов и выживаемость при использовании 5-азацитидина оказались значительно лучше [31], чем при применении низкодозовой ХТ [23, 33–35]. Так, по опубликованным данным [35], при использовании интенсивной ХТ ПР развивалась у 46 % пациентов с ОМЛ/МДС при МПН, но продолжительность ее была короткой, а медиана свободной от прогрессии выживаемости составила только 5 месяцев. В то же время при использовании для лечения 5-азацитидина [31] медиана длительности ответа колебалась от 9 до 20 месяцев. Медиана выживаемости, по данным [31], составила 11 месяцев, тогда как при проведении ХТ или симптоматической терапии — всего 2,6–5 месяцев [9, 34, 35].
В исследовании применения 5-азацитидина при первичном или развившемся после ЭТ/ИП миелофиброзе [30] четверо из семи пациентов с ИМФ в стадии трансформации ответили на лечение 5-азацитидином. Медиана выживаемости составляла 8 месяцев, тогда как при применении ХТ медиана выживаемости была значительно короче [29, 30].
Эти и другие приведенные выше данные свидетельствуют о том, что 5-азацитидин у пациентов с МПН с трансформацией ОМЛ/МДС может возвратить заболевание в хроническую фазу быстрее, чем ХТ с супрессией опухолевого клона. Подобный механизм действия азацитидина на ген гипометилирования [36, 37] был предположен de novo при МДС. Исследование статуса отдельных генов при ИМФ выявило гиперметилирование в calcitonin gene 5′ зоне [40], а также гена RARbeta2 у 90 % пациентов [25]. Анализ метилирования промотерных регионов генов p14ARF, p15INK4b, p16INK4a, Rb, hMLH1, hMSH2, APC и DAPK у 30 пациентов с ИМФ в развернутой фазе заболевания установил отсутствие процесса метилирования [20], тогда как гиперметилирование генов p15INK4 и p16INK4a часто наблюдается у пациентов с ИМФ при трансформации в ОМЛ [20].
В первых исследованиях по применению 5-азацитидина при трансформации МПН [29–31] авторы использовали дозы, рекомендованные для лечения пациентов с МДС. Большинство ответов были, к сожалению, короткими, в связи с чем было высказано мнение, что необходима консолидация терапии с применением аллогенной ТКМ или комбинирование с другими цитостатическими препаратами [29–31].
Выводы
Таким образом, данные литературы по результатам применения 5-азацитидина при трансформации МПН в ОМЛ/МДС показали, что этот гипометилирующий препарат может индуцировать ответ с низкой токсичностью у большинства пациентов [35]. Наш небольшой собственный опыт применения 5-азацитидина (Виндуза, компания «Др. Реддис Лабораториз») у пациента с бластной трансформацией ИМФ также свидетельствует о высокой эффективности препарата и его низкой токсичности.
Конфликт интересов. Не заявлен.
Список литературы
1. Berk P.D., Goldberg J.D., Silverstein M.N. et al. Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N. Engl. J. Med. 1981. 304 (8). 441-447.
2. Finazzi G., Caruso V., Marchioli R. et al. Acute leukemia in polycythemia vera. an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005. 105 (7). 2664-2670.
3. Kiladjian J.J., Rain J.D., Bernard J.F. et al. Long-term incidence of hematological evolution in three French prospective studies of hydroxyurea and pipobroman in polycythemia vera and essential thrombocythemia. Semin. Thromb. Hemost. 2006. 32 (4 Pt 2).
417-421.
4. Kiladjian J.-J., Chevret S., Abgrall J.-F. et al. Risk stratification for survival and clonal progression in essential thrombocythemia (ET): result of a prospective study of 108 patients with very long term follow up [abstract]. Blood. 2008. 112 (11). Abstr. 1747.
5. Capello D., Deambrogi C., Rossi D. et al. Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia-negative chronic myeloproliferative disorders. Br. J. Haematol. 2008. 141 (4). 504-511.
6. Wang J.C., Chen W., Nallusamy S. et al. Hypermethylation of the P15INK4b and P16INK4a in agnogenic myeloid metaplasia (AMM) and AMM in leukaemic transformation. Br. J. Haematol. 2002. 116 (3). 582-586.
7. Jost E., do ON, Dahl E. et al. Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia. 2007. 21 (3). 505-510.
8. Fourouclas N., Li J., Gilby D.C. et al. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica. 2008. 93 (11). 1635-1644.
9. Passamonti F., Rumi E., Arcaini L. et al. Leukemic transformation of polycythemia vera. a single center study of 23 patients. Cancer. 2005. 104 (5). 1032-1036.
10. Tam C.S., Kantarjian H., Cortes J. et al. Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J. Clin. Oncol. 2009. 27 (33). 5587-5593.
11. Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007. 128. 683-692.
12. Martyre M.C. Critical review of pathogenetic mechanisms in myelofibrosis with myeloid metaplasia. Curr. Hematol. Rep. 2003. 2. 257-263.
13. James C., Ugo V., Le Couedic J.P. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005. 434. 1144-1188.
14. Baxter E.J., Scott L.M., Campbell P.J. et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005. 365. 1054-1061.
15. Kralovics R., Passamonti F., Buser A.S. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005. 352. 1779-1790.
16. Levine R.L., Wadleigh M., Cools J. et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer. Cell. 2005. 7. 387-397.
17. Steensma D.P., Dewald G.W., Lasho T.L. et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both "atypical" myeloproliferative disorders and the myelodysplastic syndrome. Blood. 2005. 106. 1207-1209.
18. Jones A.V., Kreil S., Zoi K. et al. Widespread occurrence of the JAK2 V617 mutation in chronic myeloproliferative disorders. Blood. 2005. 106. 2162-2168.
19. Wolanskyj A.P., Lasho T.L., Schwager S.M. et al. JAK2 mutation in essential thrombocythaemia. clinical associations and long-term prognostic relevance. Br. J. Haematol. 2005. 131. 208-213.
20. Campbell P.J., Griesshammer M., Dohner K. et al. V617F mutation in JAK2 is associated with poorer survival in idiopathic myelofibrosis. Blood. 2006. 107. 2098-2100.
21. Prchal J.T. Myeloproliferative diseases revealed. the molecular basis and potential for targeted therapy of polycithemia vera, idi–opathic myelofibrosis, and essential thrombocythemia. Program and abstracts of the 2005 Am. Soc. of Hematol. Ann.l Meeting and Exposition. Dec. 10-13, 2005. Atlanta, Georgia. Special Plenary Session.
22. Harrison C., Kiladjian J.J., Al-Ali H.K. et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012. 36 (9). 787-798.
23. Mesa R.A. Assesing new therapies and their overall impact in myelofibrosis. Hematology Am. Soc. Hematol. Educ. Program. 2010. 2010. 115-121.
24. Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012. 119 (12). 2721-2730.
25. Mascarenhas J., Mrcado A., Rodriguez A. et al. Prolonged low dose therapy with a pan-deacetylase inhibitor, panobinostat (LBH589), in patients with myelofibrosis. Blood (ASH Annual Me–eting Abstracts). 2011. 118 (21). 794.
26. Silverman L.R., Demakos E.P., Peterson B.L. et al. Randomi–zed controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J. Clin. Oncol. 2002. 20. 2429-2440.
27. Kantarjian H., Oki Y., Garcia-Manero G. et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007. 109. 52-57.
28. Fenaux P., Mufti G.J., Hellstrom-Lindberg E. et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet оncol. 2009. 10 (3). 223-232.
29. Mesa R.A., Verstovsek S., Rivera C. et al. 5-Azacitidine has limi–ted therapeutic activity in myelofibrosis. Leukemia. 2009. 23 (1). 180-182.
30. Quintás-Cardama A., Tong W., Kantarjian H. et al. A phase II study of 5-azacitidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia. 2008. 22 (5). 965-970.
31. Thepot S., Itzykson R., Seegers V. et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010. 116 (19). 3735-3742.
32. Tefferi A., Barosi G., Mesa R.A. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT). Blood. 2006. 108. 1497-1503.
33. Cervantes F., Tassies D., Salgado C. et al. Acute transformation in nonleukemic chronic myeloproliferative disorders. actuarial probability and main characteristics in a series of 218 patients. Acta Haematol. 1991. 85 (3). 124-127.
34. Mesa R.A., Li C.Y., Ketterling R.P. et al. Leukemic transformation in myelofibrosis with myeloid metaplasia. asingle-institution experience with 91 cases. Blood. 2005. 105 (3). 973-977.
35. Tam C.S., Nussenzveig R.M., Popat U. et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood. 2008. 112 (5). 1628-1637.
36. Baylin S.B., Ohm J.E. Epigenetic gene silencing in cancer — a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer. 2006. 6 (2). 107-116.
37. Yu L., Liu C., Vandeusen J. et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat. Genet. 2005. 37 (3). 265-274.
39. Bogani C., Ponziani V., Guglielmelli P. et al. Hypermethy–lation of CXCR4 promoter in CD34cells from patients with primary myelofibrosis. Stem. Cells. 2008. 26 (8). 1920-1930.
40. Ihalainen J., Juvonen E., Savolainen E.R., Ruutu T., Palotie A. Calcitonin gene methylation in chronic myeloproliferative disorders. Leukemia. 1994. 8. 230-235.

/25-1.jpg)
/26-1.jpg)
/27-1.jpg)