Останнім часом багато дослідників розглядають метаболічні/енергетичні порушення не тільки як наслідок, але і як одну з базових причин розвитку нейродегенеративної патології центральної нервової системи (ЦНС) та пов’язаних із нейродегенерацією когнітив-них порушень, загальновідомим атрибутом яких є прогресуюча втрата певних типів нейронів. Незважаючи на те, що за причинами виникнення нейродегенеративні зміни можуть бути поліетіологічними, у них є цілий спектр спільних молекулярних і клітинних патофізіологічних процесів. Це такі процеси, як агрегація білка, глутаматна токсичність, кальцієвий, протеолітичний та окиснювальний стрес, нейрозапалення й мітохондріальна дисфункція. Вважається, що мітохондріальна дисфункція в широкому сенсі (на противагу первинній дисфункції, що пов’язана з генетичними порушеннями структурних і функціональних мітохондріальних одиниць і складових) є досить типовим і деякою мірою універсальним патологічним процесом. Мітохондрія в нейроні є ключовою клітинною органелою, що забезпечує належний рівень якості життя й функціонування нервової клітини. Слід зазначити, що мітохондріальна дисфункція при нейродегенеративних станах може мати як загальні спільні риси: пошкодження різних ланок циклу Кребса, дихального ланцюга, бета-окиснення тощо, так і певні специфічні порушення, притаманні окремій нейродегенеративній нозології. Вивчення порушень цих процесів допомагає встановити терапевтичні мішені з метою корекції вищевказаних патологічних станів за допомогою вже відомих фармакологічних засобів і для розробки нових лікарських препаратів.
Важливім фактором є те, що нейродегенеративні захворювання є залежними від віку і їх розвиток тісно пов’язаний із загальними механізмами старіння нервової системи. І своєчасна діагностика когнітивних розладів є перспективною в плані призначення адекватного лікування, що уповільнює прогресування цих порушень. Поряд із класичними уявленнями про амілоїдо-/таупатії і синуклеїнопатії як про головні патологічні прояви нейродегенерації останнім часом з’являються й альтернативні гіпотези.
Однією з таких теорій загального розвитку нейродегенерації є гіпотеза мітохондріальної дисфункції як основного патологічного процесу, що якраз і спричиняє нейродегенеративні зміни/стани ЦНС [1–3]. Автори в пошуках інтегративної теорії нейродегенеративних захворювань висувають гіпотезу, що метаболічний/енергетичний дефіцит у певних вразливих нейронних кластерах ЦНС є загальною основою, що пов’язує багато аспектів цих захворювань і може пояснити подальший розвиток патологічного процесу. Вразливі нейрональні кластери — це первинні центри розвитку нейродегенерації в ЦНС.
При кожному з видів нейродегенеративної патології нейронні кластери є специфічними, але завдяки тому, що дані регіони мають високі енергетичні потреби й багаті на синапатичні зв’язки, будь-яке погіршення енергетичного забезпечення даних зон може призводити до розвитку нейродегенерації. Іншими словами, будь-який метаболічний збій у цих нейронах викликає ланцюг подій, що спричиняють розвиток різних проявів нейродегенеративної патології. Автори підкреслюють, що саме вибіркова вразливість специфічних ней-ронних скупчень на ранніх стадіях розвитку хвороби з подальшим поширенням захворювання на вищі кіркові ділянки є типовою схемою прогресування захворювання. Метаболічний/енергетичний дефіцит також впливає на судинні й гліальні реакції, що, у свою чергу, теж впливає на процеси нейродегерації. На думку авторів даної гіпотези, вона допоможе зрозуміти основний механізм розвитку багатьох аспектів нейродегенерації і може сприяти розробці більш перспективних терапевтичних втручань у майбутньому з огляду на те, що на сьогодні дія сучасних терапевтичних засобів загалом має симптоматичний характер.
Схематично теорія метаболічного/енергетичного дефіциту, що, на думку її авторів, є загальною для нейродегенерації, виглядає так. Сукупність первинних чинників нейродегенерації починає негативно вливати на цільові нейрональні кластери. Для підтримки структурної і функціональної цілісності популяція цих нейронних кластерів намагається зберегти велику кількість синаптичних зв’язків для виконання специфічних функцій, які, як відомо, пов’язані з підвищеною потребою в енергії. Через підвищений попит на енергію мітохондрії мають тенденцію виробляти надмірну кількість активних форм кисню (АФК), що згодом впливає на морфологію мітохондрій і призводить до метаболічного стресу. Порушення функції транспортера глутамату в гліальних клітинах призводить до надмірного його накопичення в синаптичній щілині й активує постсинаптичні AMPA-/NMDA-рецептори. Це, у свою чергу, веде до збільшення надходження кальцію в постсинаптичних нейронах, порушуючи його гомеостаз в мітохондріях та ендоплазматичному ретикулумі, і знов-таки викликає надмірний рівень генерації АФК. Надмірна кількість АФК не може адекватно нівелюватись внутрішньою системою антиоксидантного захисту. АФК, з одного боку, спричиняють прямі руйнівні впливи й через метаболічний стрес, дегенеруючи нейрони, виділяють запальні цитокіни, які можуть розпізнаватися астроцитами й мікроглією. Підвищена запальна відповідь також може призводити до надмірного утворення АФК. Отже, на різних метаболічних шляхах замикаються патологічні порочні кола. Сигнали запалення й сигнали, пов’язані із АФК, можуть викликати зміни в ендотеліальних клітинах і перицитах у мозковому судинному бар’єрі, що приводить до підвищення проникності мембрани, порушення її цілісності. Це, у свою чергу, сприяє появі в мозку речовин, які в нормі не мають там бути. Усі вищеперераховані події додатково підсилюють загальну запальну відповідь, порушують цілісність судин і призводять до зменшення регіонального мозкового кровотоку. Втрата нормального зв’язку між глією і судинними системами призводить до явища інсулінорезистентності, яка додатково викликає запалення й знижує поглинання глюкози нервовими клітинами, що призводить до гіпометаболізму глюкози в мозку. Крім того, знижений рівень глюкози впливає і на лактатний човник, що, у свою чергу, призводить до зниження окисного фосфорилювання й спричиняє недостатність енергетичного забезпечення нейронів і подальший розвиток нейродегенеративних процесів.
Мітохондріальна дисфункція при нейродегенеративній патології
Разом з наявністю загальних шляхів реалізації метаболічного/енергетичного дефіциту й загальної мітохондріальної дисфункції окремі нозологічні нейродегенеративні порушення характеризуються певними специфічними змінами в енергетичному забезпеченні й роботі мітохондріальної системи.
При хворобі Альцгеймера (ХА) розвиток мітохондріальної дисфункції пов’язаний з багатьма ланками функціонування мітохондріального й синаптичного апарату нейронів. Причому шляхи розгортання метаболічного/енергетичного дефіциту можуть як мати спільні риси, так і відрізнятися від загальновизнаного патологічного шляху, пов’язаного з амілоїдогенезом.
Реалізація когнітивних функцій тісно пов’язана з адекватною роботою синапсів. Зменшення кількості та якості роботи синаптичного апарату пов’язане з розвитком і проявами когнітивного дефіциту при ХА. Синаптична комунікація в нервовій системі є надзвичайно енергоємним процесом, що регулюється кальцієвими сигналами. Мітохондрії не тільки забезпечують енергію для реалізації синаптичної функції, але й виступають як потужний кальцієвий буфер. Пресинаптичні мітохондрії забезпечують можливість тривалої синаптичної активності (саме тому вони активно транспортуються в пресинаптичну зону нейронів). Мітохондрії здатні бути потужним буфером для пресинаптичних кальцієвих сигналів, тим самим модулюючи нейротрансмісію і потенційно лімітуючи її верхню межу й максимальну потужність. Кальцієві сигнали також запускають процес злиття синаптичних пухирців в активній зоні пресинаптичної мембрани, спорожнюючи їх, так що нейротрансмітерний вміст потрапляє в синаптичну щілину. У свою чергу, нейротрансмітер впливає на постсинаптичні рецептори, приводячи до передачі сигналу на постсинаптичний нейрон. З огляду на вищенаведене мітохондріальна активність пресинаптичних нейронів забезпечує належне функціонування синаптичної нейротрансмісії, тому будь-які порушення роботи пресинаптичних мітохондрій можуть сприяти розвитку нейродегенерації саме за рахунок порушення синаптичного гомеостазу. Окрім того, порушення стабільності тау-протеїну через його гіперфосфорилювання при розвитку хвороби Альцгеймера призводить до порушень транспорту мітохондрій у пресинаптичну зону нейронів, який здійснюється по мікротрубочках, що, у свою чергу, складаються з тау-протеїну. Фосфорильований тау-білок пригнічує антероградний рух мітохондрій у нейронах.
Відомо, що агрегація β-амілоїдного пептиду (Aβ) й позаклітинне відкладання амілоїдних бляшок є однією з основних патоморфологічних характеристик хвороби Альцгеймера. У контексті розвитку мітохондріальної дисфункції при ХА показано, що олігомери Aβ можуть негативно впливати на пов’язану з мітохондріями мембрану ендоплазматичного ретикулуму, що, у свою чергу, призводить до перевантаження мітохондрій кальцієм і наступного запуску патологічних внутрішньоклітинних патологічних каскадів.
Також було продемонстровано, що Аβ і гіперфосфориляція тау-протеїну призводять до порушення процесу мітохондріальної динаміки (синтезу й поділу), мітофагії та всієї системи, що пов’язана з роботою мітохондрій у нервовій клітині [4, 5].
Активні форми кисню, що утворюються у великих кількостях при розвитку мітохондріальної дисфункції, призводять до структурних порушень мітохондріальних і клітинних мембран і до негативних наслідків роботи синаптичного апарату нервових клітин. Щодо холінергічних нейронів, які в першу чергу страждають при хворобі Альцгеймера, то певна роль у розвитку мітохондріальної дисфункції в них належить молекулам L-карнітину (ЛКАР) та ацетил-L-карнітину (Aцетил-ЛKAP). Зміни метаболізму ЛКАР/Ацетил-ЛКАР призводять до зменшення надійності функціонування мітохондріальної системи не тільки через зменшення виробництва енергії та очищення токсичних скупчень жирних кислот у мітохондріях, але й через здатність бути попередником ацетилхоліну й стабілізатором мітохондріальних мембран.
Специфічні прояви мітохондріальної дисфункції при хворобі Паркінсона, що включають, зокрема, надекспресію, неправильне складання й агрегацію α-синуклеїну, пригнічують перший мітохондріальний комплекс ферментів і деполяризують мітохондріальну мембрану, призводячи до її подальшої фрагментації. Наслідком цього є зменшення вироблення енергії органелами. Надекспресія α-синуклеїну також призводить до уповільнення виділення дофаміну в синаптичну щілину. Мутації в генах PINK1 і PARK2 також сприяють порушенню належного маркування мітохондрій для мітофагів для вчасної їх утилізації, оскільки збільшення числа неефективно працюючих органел потенційно веде не тільки до зменшення генерації енергії, але й, з великою вірогідністю, до запуску апоптозу дофамінергічних нейронів [6]. Отже, поєднання загальних і специфічних процесів, що сприяють розвитку мітохондріальної дисфункції при найбільш поширених формах нейродегенеративної патології, становить певний інтерес у контексті пошуків можливих терапевтичних впливів, у тому числі направлених на уповільнення розвитку захворювання, і дослідження потенційних терапевтичних агентів для впливу на мітохондріальну дисфункцію при нейродегенеративній патології. Певний інтерес у даному контексті становлять ендогенні молекули й сполуки. Однією з них є молекула L-карнітину.
Мітохондрії як терапевтична мішень при нейродегенерації, роль карнітину
L-карнітин — це природна сполука, наявна в майже всіх тканинах ссавців, у тому числі в мозку. З їжею карнітин надходить в організм (переважно він наявний у червоному м’ясі й молочних продуктах), а також синтезується в нирках, печінці й мозку [1]. L-карнітин вважається умовно необхідною поживною речовиною для людини, особливо за певних обставин, коли можуть спостерігатись його низькі внутрішньоклітинні рівні. Дефіцит L-карнітину може спостерігатись у недоношених дітей, літніх людей, пацієнтів із цукровим діабетом, генетичними порушеннями, що призводять до первинного або вторинного карнітинового дефіциту [7, 8].
Основна й найбільш досліджена функція L-карнітину полягає в транспортуванні активованих довголанцюгових жирних кислот (довголанцюгових жирних ацил-CoA) у мітохондрії для їх подальшої деградації шляхом β-окиснення з вивільненням енергії у формі аденозинтрифосфату (АТФ) [9].
Тканини людини, включно з мозком, містять вільний L-карнітин, а також ацильовані похідні вуглецевих ланцюгів різної довжини, що включають ацетильовані й пальмітильовані похідні [9]. Існують дані щодо концентрацій різних форм метаболітів карнітину в ЦНС. Так, ізольовані нейрони мозку дорослих містять приблизно 80 % вільного L-карнітину, 10–15 % ацетил-L-карнітину й менше ніж 10 % довголанцюгових форм ацилкарнітину [10].
Ацетил-L-карнітин є одним з основних метаболітів карнітину, має досить високу біодоступність, проходить через мозковий бар’єр краще, ніж карнітин, але в мозку він за необхідності легко перетворюється на карнітин [17].
L-карнітин та ацетил-L-карнітин все частіше привертають увагу дослідників через наявність у них низки потенційних нейропротекторних властивостей [11–14, 18–20]. Існують дані про нейропротекторні ефекти L-карнітину при ураженнях центральної і периферичної нервової системи в дорослих [15, 16], його позитивні ефекти при дозріванні нервової системи та її старінні, а також при розвитку залежної від віку нейродегенеративної патології. Експериментальні дані свідчать, що ацетил-L-карнітин підсилює продукцію АТФ у мітохондріях, допомагає захистити їх від окиснювального стресу й сприяє кращій виживаності ней-ронів при моделюванні нейродегенеративної патології. Властивості молекули та її ефекти запобігають втраті мозкових функцій, що зазвичай відбувається під час старіння, а її нейропротекторні ефекти реалізуються в гіпокампі, передній зоні кори, чорній речовині й частині мускаринових рецепторів ЦНС. До них належать антиоксидантна активність, покращання функцій мітохондрій, стабілізація внутрішньоклітинних мембран і холінергічна нейротрансмісія.
Отримані дані досліджень демонструють, що введення Ацетил-ЛКАР після травми може покращити функцію мітохондрій [21], зменшити набряк мозку й запобігти втраті тканин [19, 22]. Тривалий прийом Ацетил-ЛКАР покращував енергетичний стан здорового мозку мишей [23].
Продемонстровано, що оскільки Ацетил-ЛКАР метаболізується до ацетилу-CoA, що має потенціал до ацетилювання гістонів, він може впливати на експресію генів [24]. Також він метаболізується до ацетилювання білків і ферментів, що може значно змінювати їх активність [25, 26]. У результаті внутрішньомозкового клітинного метаболізму ацетильний фрагмент з Ацетил-ЛКАР може бути окиснений для отримання енергії, може виступати попередником ацетилхоліну й включатись до амінокислотних нейромедіаторів і ліпідів мозку [27–29].
Дослідження в експериментальних патологічних моделях дають певне зрозуміння щодо можливих механізмів реалізації нейропротекції L-карнітину. Так, Y. Ueno et al. [30], вивчаючи модель хронічної гіпоперфузії головного мозку в дорослих щурів, спричиненої постійною перев’язкою обох загальних сонних артерій, показали, що щури, які отримали перорально 600 мг/кг L-карнітину після оклюзії артерії, демонстрували кращі показники когнітивної діяльності (проходження водного лабіринту Морріса) під час тестування через 28 днів після операції. Разом з покращанням когнітивних функцій група, що отримувала L-карнітин після хронічної гіпоперфузії, продемонструвала менший рівень пошкодження ДНК і перекисного окиснення ліпідів, збільшення товщини мієлінової оболонки й посилення експресії маркерів олігодендроцитів. Через 28 днів після початку гіпоперфузії в щурів, які отримали L-карнітин, спостерігались підвищення фосфорильованого Akt і рапаміцинового рецептора ссавців (mTOR), підвищення рівня фосфорильованого високомолекулярного нейрофіламенту (pNFH) порівняно з щурами, які отримували плацебо. Таким чином автори даного дослідження припустили, що в умовах гіпоперфузії мозку L-карнітин «регулює сигнальний шлях PTEN/Akt/mTOR і підвищує пластичність аксонів при одночасному зменшенні окиснювального стресу й збільшенні мієлінізації олігодендроцитів аксонів», і саме ці ефекти можуть бути відповідальними за зменшення когнітивних порушень, що виникли в результаті моделювання хронічної гіпоперфузії мозку [30].
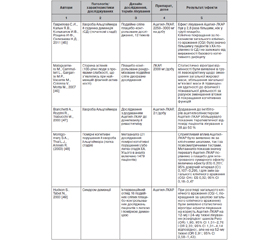
За досить тривалий час дослідження молекули ЛКАР/Ацетил-ЛКАР були встановлені численні потенційні нейропротекторні механізми реалізації даного ефекту, що наведені в табл. 1 згідно з оглядом L. Jones et al. [31].
L-карнітин/Ацетил-L-карнітин і нейродегенеративна патологія
Накопичені дані щодо низки потенційно корисних біологічних ефектів ЛКАР і його метаболітів обумовили вивчення його в контексті розвитку різноманітних нейродегенеративних порушень ЦНС. Так, низка досліджень показала, що введення Ацетил-ЛКАР підтримує і/або підвищує рівень ацетилхоліну в мозку [32]. De Simone et al. (1991) [33] виявили підвищену активність холінової ацетилтрансферази й експресію рецептора фактору росту нервів (NGF) у стріатумі, підвищений рівень білка NGF у гіпокампі щурів після введення Ацетил-ЛКАР у шлуночки мозку. Загальновідомо, що ацетилхолін відіграє важливу роль у навчанні й пам’яті [34]. Інтактні холінергічні шляхи в базальному передньому мозку та гіпокампі необхідні для нормального функціонування навчання, роботи систем уваги й пам’яті [35]. Ацетилхолін частково викликає синапатичну пластичність гіпокампа й кори за допомогою взаємодії «астроцит — нейрон» [36]. Ацетилхолін і/або холінергічна стимуляція відіграють важливу роль у захисті мозку щодо розвитку нейрозапалення, тому дефіцит даного нейромедіатора пов’язують з розвитком когнітивних порушень при нейродегенерації.
S. Furukawa et al. [37] продемонстрували, що попереднє введення щурам інгібітору ацетилхолінестерази галантаміну за 2 години до травми призводило до значного зменшення пошкодження гіпокампа й кори за 7 днів після отримання травми. Попереднє введення щурам галантаміну також призводило до зменшення посттравматичного накопичення мікроглії і зменшення кількості прозапального IL-1β у мозку. В інших дослідженнях повідомлялось, що введені карнітин та ацильний фрагмент можуть бути використані нейронами для генерації енергії, синтезу ацетилхоліну [27], демонструють холіноміметичні властивості [38], беруть участь у синтезі амінокислотних нейромедіаторів [39] і ліпідів [29]. Також було встановлено, що Aцетил-ЛКАР має протизапальну дію, приводить до стабілізації мембран [10], діє як антиоксидант, захищаючи нервові клітини від окиснювального стресу, посилює активність фактора росту нервів і може потенціювати енергетичний метаболізм. Карнітин в експерименті підвищував інгібування ацетилхолінестерази галантаміном у гіпокампі [40].
Дослідження плазмових концентрацій середньоланкових ацилкарнітинів у пацієнтів з помірними когнітивним порушеннями й ХА порівняно з когнітивно інтактними особами показали наявність значного дефіциту даних молекул у плазмі крові людей із когнітивним дефіцитом. Причому пацієнти з ХА мали найнижчі рівні середньоланкових ацилкарнітинів в плазмі серед обстежених груп, при тому що дані концентрації корелювали із зменшеними мозковими об’ємами сірої речовини префронтальної кори й наявністю когнітивного порушення [41]. Аналогічні дані щодо наявного прогресивного зниження плазмових концентрацій у контину-умі «когнітивна норма — легкі когнітивні порушення — помірні когнітивні порушення — хвороба Альцгеймера» (5,6 ± 1,3 μмоль/л — 4,3 ± 0,9 μмоль/L — 4,00 ± 0,53 μмоль/л — 3,5 ± 0,6 μмоль/л) були продемонстровані в іншому дослідженні [42]. Автори вищенаведеного дослідження пропонують розглядати сироваткові рівні ацетил-L-карнітину та інших ацил-L-карнітинів як предиктивний маркер нейродегенеративного процесу на початкових рівнях його розвитку, хоча й зазначають, що для остаточного доказу даної гіпотези знадобляться лонгітудинальні спостереження.
Хоча селективна втрата дофамінергічних нейронів і накопичення α-синуклеїну на даний час становлять основну гіпотезу, що пояснює розвиток хвороби Паркінсона, також існують докази участі в розвитку цього захворювання й інших патологічних процесів: окиснювального стресу, мітохондріальної і протеосомної дисфункції, причому останнім часом особливі ролі в патогенезі захворювання відводять саме двом останнім — як базовим дисфункціям, що відповідальні за метаболічний/енергетичний дефіцит вразливих пулів (дофамінергічних) нейронів.
В експериментальних дослідженнях при моделюванні хвороби Паркінсона (ХП) був, зокрема, продемонстрований позитивний вплив Aцетил-ЛКАР на гіпокампальний нейрогенез через інгібіцію внутрішньоклітинних сигналів, пов’язаних з апоптичною смертю клітин (GSK-3β і BAX) і, відповідно, підвищенням їх виживання через проліферацію нейронних прогеніторних клітин (NPC) і диференціювання нейронів у гіпокампі [43]. Ацетил-L-карнітин регулює мітохондріальний метаболізм і, як повідомляється, покращує когнітивні функції при різних нейродегенеративних захворюваннях. При дослідженні впливу Aцетил-ЛКАР на нейрогенез у дорослих щурів на моделі паркінсонічного фенотипу, спричиненого введенням 6-гідроксидопаміну (6-OHDA), дослідники з’ясовували можливий основний механізм дії. Одностороннє введення 6-OHDA в медіальний пучок переднього мозку знижувало проліферацію нейронних прогеніторних клітин і негативно впливало на тривалу виживаність і диференціювання нейронів у гіпокампі. Під час експерименту було показано, що хронічне введення Aцетил-ЛКАР (100 мг/кг/добу інтраперитонеально) потенційно посилювало проліферацію, довготривалу виживаність і нейрональну диференціацію NPC. Лікування ALCAR стимулювало сигнали, пов’язані з виживанням клітин, через AKT і BCL-2, інгібуючи сигнали, пов’язані зі смертю клітин, через GSK-3β і BAX. Отже, автори на основі отриманих даних роблять висновок, що Aцетил-ЛКАР чинить нейропротекторні ефекти проти 6-OHDA-індукованого порушення ней-рогенезу в гіпокампі, регулюючи підвищення виживаності клітин і пов’язані з апоптозом сигнали.
Дослідження Afshin-Majd et al. також продемонструвало наявність нейропротекторного ефекту Ацетил-ЛКАР у дозах 100 або 200 мг/кг/добу при моделювання ХП при внутрішньошлуночковому введенні 6-гідроксидопаміну. Захисний ефект Ацетил-ЛКАР опосередковувався через механізми гальмування ней-розапалення, апоптозу, астрогліозу й окиснювального стресу [44].
Клінічні дослідження ЛКАР/Ацетил-ЛКАР при нейродегенеративній патології
Наявність великої кількості експериментальних даних щодо багатьох потенційних нейропротекторних властивостей молекули та інших потенційних терапевтичних властивостей сприяли їх оцінці в рамках клінічних досліджень. Найбільша база клінічних досліджень L-карнітину/ацетил-L-карнітину була отримана при нейродегенеративній патології альцгеймерівського спектра (табл. 1).
Сумарний ефект Ацетил-ЛКАР у рамках впливу на помірні когнітивні порушення/легку стадію ХА, за результатами метааналізу S.A. Montgomery, L.J. Thal, R. Amrein (2003), дорівнює 0,201 [48]. Автори метааналізу окремо оцінили величину ефекту в різних психометричних тестах (близько 40), що використовувались у включених до метааналізу дослідженнях. Вони були згруповані в п’ять категорій: увага/продуктивність (10 інструментів), пам’ять (17 інструментів), навчання (3 інструменти), малювання (4 інструменти) та вищі інтелектуальні функції (6 інструментів). Показники величини ефекту для вищезгаданих тестів, виключно з малюванням і навчанням, показали значний позитивний ефект для Ацетил-ЛКАР, що становив від 0,21 до 0,37. Але водночас інтервенційний огляд подвійних сліпих плацебо-контрольованих досліджень із використанням Ацетил-ЛКАР у когорті пацієнтів із синдромом деменції, проведений S. Hudson, N. Tabet (2003) [49], демонструє набагато скромніші клінічні результати й висновки.
Слід зазначити що, на жаль, у клінічній практиці відсутній препарат, який зміг би задовольнити пацієнтів і лікарів щодо ефективності свого терапевтичного впливу на нейродегенеративну патологію. Загальноприйнятий стандарт лікування хвороби Альцгеймера з використанням інгібіторів ацетилхолінестерази демонструє величину ефекту або стандартизовану середню різницю (SMD), що становить від 0,15 до 0,28 [57] і від 0,16 до 0,24 для інгібіторів ацетилхолінестерази й мемантину відповідно [58] і від 0,32 до 0,49 у більш ранніх дослідженнях для мемантину [59]. Дещо кращі показники отримані при комбінованій терапії двома групами препаратів, але разом з тим клініцистам добре відома їх ефективність у реальному житті. Більше того, існують результати досліджень щодо відсотка респондерів при лікування ХА інгібіторами ацетилхолінестерази й мемантином серед клінічної популяції хворих [60, 61].
Отже, у низці досліджень карнітину і його активних метаболітів одержали певні цікаві клінічні результати.
Так, обсерваційні дослідження показали, що пацієнти з когнітивними порушеннями, починаючи з найбільш легких форм, мають порівняно з контролем значно нижчі плазмові концентрації ЛКАР, що продовжують знижуватись при наростанні ступеня тяжкості когнітивного дефіциту в континуумі «легкі — помірні когнітивні порушення — деменція». З одного боку, даний факт потенційно може бути використаний як предиктор розвитку (ранній маркер) формування ней-рокогнітивного захворювання, а з іншого — для постановки питання про можливе використання молекули в комплексній терапії.
Додавання Ацетил-ЛКАР до стандартної терапії інгібіторами ацетилхолінестерази збільшувало клінічну ефективність останніх з 38 до 50 %, що теж має значення в підвищенні ефективності терапії захворювання [47].
Одним з досить поширених клінічних симптомів при розвитку ХА та ХП є синдром астенії, і стандартні лікарські засоби, що використовуються за протоколом лікування, не досить ефективні щодо його корекції. Слід зауважити, що клінічно астенія має як когнітивний/центральний, так і фізичний (соматичний) компоненти й загалом не сприяє покращанню якості життя людей з нейродегенеративною патологією. Можливий внесок у розвиток астенії роблять наростаюча мітохондріальна дисфункція і, безумовно, процес старіння, причому не тільки нервової системи, але й усього організму в цілому. У даному контексті досить цікаво виглядають дослідження карнітину з включенням у дослідження осіб старечого віку (100 років) із симптомами вираженої астенії. Його результати показали досить позитивні ефекти ЛКАР як щодо фізичної, так і щодо психічної втоми, що також призводили до покращання когнітивної діяльності таких пацієнтів [55].
Незважаючи на досить великий обсяг експериментальних досліджень з використанням моделей ХП, клінічні дослідження ЛКАР/Ацетил-ЛКАР при ХП, які вдалось відшукати в базі даних PubMed, були обмежені тільки обсерваційними спостереженнями, причому з прямо протилежними результатами. Так, дослідження рівня ЛКАР у спинномозковій рідині хворих на ХП [62] не показало наявності різниці в концентрації ЛКАР у хворих на ХП і в контрольній групі. Однак інше дослідження, проведене у 2017 році, продемонструвало протилежні результати. Його результати показали, що концентрація ЛКАР досить добре може відмежовувати групу контролю і хворих, які знаходяться на початковій стадії розвитку ХП, а молекула карнітину може розглядатись як можливий маркер розвитку ХП. Дослідники [63] показали, що статистично вірогідне зниження рівня п’яти довголанцюгових ацилкарнітинів було виявлене у хворих на ХП І стадії за Хен — Яром. Аналогічно до зниження рівня ацилкарнітину зниження відношення ацилкарнітину до жирної кислоти (16 : 0) і підвищений індекс карнітинової пальмітоїлтрансферази 1-го типу було виявлено у хворих уже на I стадії захворювання, що свідчить про початкове пригнічення процесу β-окиснення. Отримані дані з використанням 12–14-довголанцюгових ацилкарнітинів забезпечили велику площу під кривою, що свідчить про високий рівень специфічності й помірний рівень чутливості даного тесту при діагностиці хвороби Паркінсона за допомогою дослідженого маркера.
Останніми роками в клінічних дослідженнях ХП значну увагу приділяють так званим немоторним проявам захворювання. Це обумовлено не тільки їх внеском у загальну якість життя хворих, але й тим, що багато з них передують появі моторних симптомів і можуть бути ранніми маркерами хвороби. У цьому контексті досить цікавими є дослідження впливу карнітину й ацетилкарнітину на депресивні симптоми, що також можуть бути присутніми в пацієнтів із нейродегенерацією. Результати метааналізу, проведеного з метою оцінки терапевтичного впливу Ацетил-ЛКАР на депресивну симптоматику [64], продемонстрували доволі позитивні ефекти молекули, що були порівнянними з ефектами різних молекул антидепресантів при менший вираженості побічних явищ. Існує експериментальне дослідження щодо можливого механізму ранньої терапевтичної дії даної молекули на редукцію депресивних проявів [65].
Можливо, що нові накопичені дані, у першу чергу експериментальні, щодо мітохондріальної дисфункції при хворобі Паркінсона будуть спонукати клінічних дослідників оцінити можливості ЛКАР у комплексному лікуванні пацієнтів із ХП, особливо звертаючи увагу на вплив молекули на стан когнітивних функцій і немоторні симптоми.
Висновки
З огляду на велику кількість фундаментальних, преклінічних і клінічних досліджень стає зрозумілим, що мітохондріальна дисфункція при нейродегенеративній патології центральної нервової системи є одним з основних патофізіологічних процесів. Вона може лежати в основі багатьох клінічних феноменів. Мітохондріальна дисфункція може бути досить ефективною мішенню, на яку буде направлена нова терапія як хвороби Альцгеймера, так і хвороби Паркінсона. Пошук нових препаратів, що ефективно впливають на мітохондріальну дисфункцію при нейродегенерації, триває і дає певну надію, що вони посядуть гідне місце в лікуванні хвороби Альцгеймера й хвороби Паркінсона. L-карнітин у контексті розвитку нейродегенерації, безумовно, становить інтерес у патофізіологічному, діагностичному й терапевтичному сенсі. На сьогодні на фармацевтичному ринку України існує лікарський засіб — препарат L-карнітину, представлений під назвою Метакартин (виробник — компанія World Medicine).
Список литературы
1. Belenguer P., Duarte J.M.N., Schuck P.F., Ferreira G.C. Mitochondria and the Brain: Bioenergetics and Beyond. Neurotox. Res. 2019. 36(2). 219-238. doi:10.1007/s12640-019-00061-7.
2. Grimm A., Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 2017. 143(4). 418-431. doi:10.1111/jnc.14037.
3. Muddapu V.R., Dharshini S.A.P., Chakravarthy V.S., Gromiha M.M. Neurodegenerative Diseases — Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020. 14. 213. Published 2020 Mar 31. doi:10.3389/fnins.2020.00213.
4. Reddy P.H., Oliver D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Di-sease. Cells. 2019. 8(5). 488. Published 2019 May 22. doi:10.3390/cells8050488.
5. Вertholet A.M., Delerue T., Millet A.M. et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016. 90. 3-19. doi:10.1016/j.nbd.2015.10.011.
6. Bose A., Beal M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016. 139 Suppl 1. 216-231. doi:10.1111/jnc.13731.
7. Marcovina S.M., Sirtori C., Peracino A., Gheorghiade M., Borum P., Remuzzi G., Ardehali H. Translating the basic knowledge of mitochondrial functions to metabolic therapy: role of l-carnitine. Transl. Res. 2013. 161. 73-84.
8. Ribas G.S., Vargas C.R., Wajner M. L-сarnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene. 2014. 533. 469-476.
9. Jones L.L., McDonald D.A., Borum P.R. Acylcarnitines: role in brain. Prog. Lipid Res. 2010. 49. 61-75.
10. Carnitine and acetyl-L-carnitine roles and neuroprotection in developing brain. Neurochem. Res. Author manuscript; available in PMC 2018 Jun 1. Published in final edited form as: Neurochem. Res. 2017 Jun. 42(6). 1661-1675. Published online 2017 May 16. doi: 10.1007/s11064-017-2288-7.
11. Ferreira G.C., McKenna M.C. L-сarnitine and Acetyl-L-carnitine Roles and Neuroprotection in Developing Brain. Neurochem. Res. 2017. 42(6). 1661-1675. doi:10.1007/s11064-017-2288-7.
12. Burks S., Raymick J., Robinson B., Hanig J., Sarkar S. Neuroprotective effects of acetyl-l-carnitine (ALC) in a chronic MPTP-induced Parkinson’s disease mouse model: Endothelial and microglial effects. Neurosci Lett. 2019. 703. 86-95. doi:10.1016/j.neulet.2019.03.015.
13. Kazak F., Yarim G.F. Neuroprotective effects of acetyl-l-carnitine on lipopolysaccharide-induced neuroinflammation in mice: Involvement of brain-derived neurotrophic factor. Neurosci Lett. 2017. 658. 32-36. doi:10.1016/j.neulet.2017.07.059.
14. Bak S.W., Choi H., Park H.H. et al. Neuroprotective Effects of Acetyl-L-Carnitine Against Oxygen-Glucose Deprivation-Induced Neural. Stem. Cell Death. Mol. Neurobiol. 2016. 53(10). 6644-6652. doi:10.1007/s12035-015-9563.
15. Rolim L.C., da Silva E.M., Flumignan R.L., Abreu M.M., Dib S.A. Acetyl-L-carnitine for the treatment of diabetic peripheral neuropathy. Cochrane Database Syst. Rev. 2019. 6(6). CD011265. Published 2019 Jun 15. doi:10.1002/14651858.CD011265.pub2.
16. Cruccu G., Di Stefano G., Fattapposta F. et al. L-Acetyl-carnitine in Patients with Carpal Tunnel Syndrome: Effects on Nerve Protection, Hand Function and Pain [published correction appears in CNS Drugs. 2018 Feb 13]. CNS Drugs. 2017. 31(12). 1103-1111.
17. Bieber L.L. Carnitine. Annu Rev. Biochem. 1988. 57. 261-83.
18. Ishii T., Shimpo Y., Matsuoka Y., Kinoshita K. Anti-apoptotic effect of acetyl-l-carnitine and I-carnitine in primary cultured neurons. Jpn J. Pharmacol. 2000 Jun. 83(2). 119-24.
19. Xu S., Waddell J., Zhu W., Shi D., Marshall A.D., McKenna M.C., Gullapalli R.P. In vivo longitudinal proton magnetic resonance spectroscopy on neonatal hypoxic-ischemic rat brain injury: Neuroprotective effects of acetyl-L-carnitine. Magn. Reson. Med. 2015 Dec. 74(6). 1530-42.
20. Hota K.B., Hota S.K., Chaurasia O.P., Singh S.B. Acetyl-L-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus. 2012 Apr. 22(4). 723-36.
21. Demarest T.G., Schuh R.A., Waddell J., McKenna M.C., Fiskum G. Sex-dependent mitochondrial respiratory impairment and oxidative stress in a rat model of neonatal hypoxic-ischemic encephalopathy. J. Neurochem. 2016 Jun. 137(5). 714-29.
22. Tang S., Xu S., Lu X., Gullapalli R.P., McKenna M.C., Waddell J. Neuroprotective Effects of Acetyl-L-Carnitine on Neonatal Hypoxia Ische-mia-Induced Brain Injury in Rats. Dev. Neurosci. 2016. 38(5). 384-396.
23. Smeland O.B., Meisingset T.W., Borges K., Sonnewald U. Chronic acetyl-L-carnitine alters brain energy metabolism and increases noradrenaline and serotonin content in healthy mice. Neurochem. Int. 2012 Jul. 61(1). 100-7
24. Madiraju P., Pande S.V., Prentki M., Madiraju S.R. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics. 2009 Aug 16. 4(6). 399-403.
25. McKenna M.C., Ferreira G.C. Enzyme Complexes Important for the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016. 13. 59-98.
26. Gibson G.E., Xu H., Chen H.L., Chen W., Denton T.T., Zhang S. Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J Neurochem. 2015 Jul. 134(1). 86-96.
27. Scates P.W. Acetyl-L-carnitine as a precursor of acetylcholine. Neurochem. Res. 1990. 15. 597-601.
28. Scafidi S., Fiskum G., Lindauer S.L., Bamford P., Shi D., Hopkins I., McKenna M.C. Metabolism of acetyl-L-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J. Neurochem. 2010 Aug. 114(3). 820-31.
29. Ricciolini R., Scalibastri M., Kelleher J.K., Carminati P., Calvani M., Arduini A. Role of acetyl-L-carnitine in rat brain lipogenesis: implications for polyunsaturated fatty acid biosynthesis. J. Neurochem. 1998 Dec. 71(6). 2510-7.
30. Ueno Y., Koike M., Shimada Y., Shimura H., Hira K., Tana-ka R., Uchiyama Y., Hattori N., Urabe T. L-carnitine enhances axonal plasticity and improves white-matter lesions after chronic hypoperfusion in rat brain. J. Cereb. Blood Flow Metab. 2015. 35. 382-91.
31. Jones L.L., McDonald D.A., Borum P.R. Acylcarnitines: role in brain. Prog. Lipid. Res. 2010. 49(1). 61-75. doi:10.1016/j.plipres.2009.08.004.
32. Imperato A., Ramacci M.T., Angelucci L. Acetyl-L-carnitine enhances acetylcholine release in the striatum and hippocampus of awake freely moving rats. Neurosci Lett. 1989. 107. 251-5.
33. De Simone R., Ramacci M.T., Aloe L. Effect of acetyl-L-carnitine on forebrain cholinergic neurons of developing rats. Int. J. Dev. Neurosci. 1991. 9. 39-46
34. Gold P.E. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol. Learn. Mem. 2003. 80. 194-210; Mitsushima D., Sano A., Takahashi T. A cholinergic trigger drives learning-induced plasticity at hippocampal synapses. Nat. Commun. 2013. 4. 2760.
35. Sarter M., Bruno J.P., Givens B. Attentional functions of cortical cholinergic inputs: what does it mean for learning and memory? Neurobiol. Learn. Mem. 2003. 80. 245-56.
36. Phillis J.W. Acetylcholine release from the central nervous system: a 50-year retrospective. Crit. Rev. Neurobiol. 2005. 17. 161-217.
37. Furukawa S., Yang L., Sameshima H. Galantamine, an acetylcholinesterase inhibitor, reduces brain damage induced by hypoxia-ischemia in newborn rats. Int. J. Dev. Neurosci. 2014. 37. 52-7.
38. Janiri L., Falcone M., Persico A., Tempesta E. Activity of L-carnitine and L-acetylcarnitine on cholinoceptive neocortical neurons of the rat in vivo. J. Neural. Transm. Gen. Sect. 1991. 86. 135-46.
39. Scafidi S., Fiskum G., Lindauer S.L., Bamford P., Shi D., Hopkins I., McKenna M.C. Metabolism of acetyl-L-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J. Neurochem. 2010. 114. 820-831.
40. Svoboda Z., Kvĕtina J., Herink J. et al. Galantamine antiacetylcholinesterase activity in rat brain influenced by L-carnitine. Biomed Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2005. 149(2). 335-337.
41. Ciavardelli D., Piras F., Consalvo A. et al. Medium-chain plasma acylcarnitines, ketone levels, cognition, and gray matter vo-lumes in healthy elderly, mildly cognitively impaired, or Alzheimer’s disease subjects. Neurobiol. Aging. 2016. 43. 1-12. doi:10.1016/j.neurobiolaging.2016.03.005.
42. Cristofano A., Sapere N., La Marca G. et al. Serum Levels of Acyl-Carnitines along the Continuum from Normal to Alzheimer’s Dementia. PLoS One. 2016. 11(5). e0155694. Published 2016 May 19. doi:10.1371/journal.pone.0155694.
43. Singh S., Mishra A., Mishra S.K., Shukla S. ALCAR promote adult hippocampal neurogenesis by regulating cell-survival and cell death-related signals in rat model of Parkinson’s disease like-phenotypes. Neurochem. Int. 2017 Sep. 108. 388-396. doi: 10.1016/j.neuint.2017.05.017. Epub 2017 May 31. PMID: 28577987. DOI: 10.1016/j.neuint.2017.05.017.
44. Afshin-Majd S., Bashiri K., Kiasalari Z., Baluchnejadmojarad T., Sedaghat R., Roghani M. Acetyl-l-carnitine protects dopaminergic nigrostriatal pathway in 6-hydroxydopamine-induced model of Parkinson’s disease in the rat. Biomed. Pharmacother. 2017 May. 89. 1-9. doi: 10.1016/j.biopha.2017.02.007. Epub 2017 Feb 12.
45. Гаврилова С.И., Калын Я.В., Колыхалов И.В., Рощина И.Ф., Селезнева Н.Д. Ацетил-L-карнитин (карницетин) в лечении начальных стадий болезни Альцгеймера и сосудистой деменции. Журнал неврологии и психиатрии имени С.С. Корсакова. 2011. 111(9). 16-22.
46. Cammalleri L., Gargante M.P., Vacante M., Colonna V., Motta M. L-Carnitine treatment reduces severity of physical and mental fatigue and increases cognitive functions in centenarians: a randomized and controlled clinical trial. Am. J. Clin. Nutr. 2007. 86(6). 1738-1744. doi:10.1093/ajcn/86.5.1738
47. Bianchetti A., Rozzini R., Trabucchi M. Effects of acetyl-L-carnitine in Alzheimer’s disease patients unresponsive to acetylcholinesterase inhibitors. Curr. Med. Res Opin. 2003. 19(4). 350-353. doi:10.1185/030079903125001776.
48. Montgomery S.A., Thal L.J., Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carnitine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer’s disease. International Clinical Psychopharmaco-logy. 2003. 18(2). 61-71. doi:10.1097/00004850-200303000-00001.
49. Hudson S., Tabet N. Acetyl-L-carnitine for dementia. Cochrane Database Syst. Rev. 2003. 2003(2). CD003158. doi:10.1002/14651858.CD003158.
50. Thal L.J., Calvani M., Amato A., Carta A. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology. 2000. 55(6). 805-810. doi:10.1212/wnl.55.6.805.
51. Brooks J.O. 3rd, Yesavage J.A., Carta A., Bravi D. Acetyl L-carnitine slows decline in younger patients with Alzheimer’s disease: a reanalysis of a double-blind, placebo-controlled study using the trilinear approach. Int. Psychogeriatr. 1998. 10(2). 193-203. doi:10.1017/s1041610298005304.
52. Thal L.J., Carta A., Clarke W.R. et al. A 1-year multicenter placebo-controlled study of acetyl-L-carnitine in patients with Alzheimer’s disease. Neurology. 1996. 47(3). 705-711. doi:10.1212/wnl.47.3.705.
53. Sano M., Bell K., Cote L. et al. Double-blind parallel design pilot study of acetyl levocarnitine in patients with Alzheimer’s disease. Arch. Neurol. 1992. 49(11). 1137-1141. doi:10.1001/archneur.1992.00530350051019.
54. Spagnoli A., Lucca U., Menasce G. et al. Long-term acetyl-L-carnitine treatment in Alzheimer’s disease. Neurology. 1991. 41(11). 1726-1732. doi:10.1212/wnl.41.11.1726.
55. Cipolli C., Chiari G. Effetti della L-acetilcarnitina sul deterioramento mentale dell’anziano: primi risultati [Effects of L-acetylcarnitine on mental deterioration in the aged: initial results]. Clin. Ter. 1990. 132(6 Suppl.). 479-510.
56. Herrmann W.M., Dietrich B., Hiersemenzel R. Pharmaco-electroencephalographic and clinical effects of the cholinergic substance — acetyl-L-carnitine — in patients with organic brain syndrome. Int. J. Clin. Pharmacol. Res. 1990. 10(1-2). 81-84.
57. Rockwood K. Size of the treatment effect on cognition of cholinesterase inhibition in Alzheimer’s disease [published correction appears in J. Neurol. Neurosurg. Psychiatry. 2004 Jul. 75(7). 1086]. J. Neurol. Neurosurg. Psychiatry. 2004. 75(5). 677-685. doi:10.1136/jnnp.2003.029074.
58. Kishi T., Matsunaga S., Oya K., Nomura I., Ikuta T., Iwata N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimers Dis. 2017. 60(2). 401-425. doi:10.3233/JAD-170424.
59. Smith M., Wells J., Borrie M. Treatment effect size of memantine therapy in Alzheimer disease and vascular dementia. Alzheimer Dis. Assoc. Disord. 2006. 20(3). 133-137. doi:10.1097/00002093-200607000-00002.
60. Бачинська Н.Ю., Рожелюк І.Ф., Холін В.О., Пішель І.М., Леонов Ю.І. Ефективність лікування донепезилу гідрохлоридом хворих похилого віку із синдромом деменції альцгеймерівського і судинного типу залежно від поліморфізму гена апоЕ. Український неврологічний журнал. 2014. № 1. С. 107-115.
61. Холин В.А., Бачинская Н.Ю. Практические аспекты применения мемантина в лечении деменции альцгеймеровского типа. Международный неврологический журнал. 2016. № 1(79). С. 90-94.
62. Jiménez-Jiménez F.J., Rubio J.C., Molina J.A., Martín M.A., Campos Y., Benito-León J., Ortí-Pareja M., Gasalla T., Arenas J. Cerebrospinal Fluid Carnitine Levels in Patients With Parkinson’s Disease. J. Neurol. Sci. 1997 Feb 12. 145(2). 183-5. doi: 10.1016/s0022-510x(96)00259-6.
63. Saiki S., Hatano T., Fujimaki M., Ishikawa K.I., Mori A., Oji Y., Okuzumi A., Fukuhara T., Koinuma T., Imamichi Y., Nagumo M., Furuya N., Nojiri S., Amo T., Yamashiro K., Hattori N. Decreased Long-Chain Acylcarnitines From Insufficient β-Oxidation as Potential Early Diagnostic Markers for Parkinson’s Disease. Sci Rep. 2017 Aug 4. 7(1). 7328. doi: 10.1038/s41598-017-06767.
64. Veronese N., Stubbs B., Solmi M. et al. Acetyl-L-Carnitine Supplementation and the Treatment of Depressive Symptoms: A Systematic Review and Meta-Analysis. Psychosomatic Medicine. 2018. Vol. 80. P. 154-159.
65. Nasca C., Xenos D., Barone Y., Caruso A., Scaccianoce S., Matrisciano F., Battaglia G., Mathé A.A., Pittaluga A., Lionetto L., Simmaco M., Nicoletti F. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc. Natl Acad. Sci. USA. 2013 Mar 19. 110(12). 4804-9.

/71.jpg)
/72.jpg)
/73.jpg)