Рання інфантильна епілептична енцефалопатія (РІЕЕ) вперше описана японським епілептологом Shunsuke Ohtahara у 1976 р. як тяжка форма епілепсії, що вражає немовлят перших місяців життя та характеризується типовими електроенцефалографічними змінами у вигляді патерну «спалах — пригнічення» [1]. Згідно з визначенням Міжнародної протиепілептичної ліги (ILAE), епілептична енцефалопатія — це стан, при якому епілептична активність сама по собі призводить до розвитку тяжких когнітивних і поведінкових порушень у дітей більшою мірою, ніж цього можливо було очікувати від основної структурної патології (наприклад, вади розвитку кори головного мозку) [2].
РІЕЕ проявляється частими тонічними судомами або епілептичними спазмами, що розпочинаються у ранньому дитячому віці та супроводжуються специфічними змінами на ЕЕГ у вигляді патерну «спалах — пригнічення». Даний патерн формується високоамплітудними спалахами активності, що чергуються з майже плоскими фазами падіння амплітуди ритму (декременти). У близько 75 % дітей РІЕЕ в подальшому трансформується в синдром Веста, що характеризується кластерними тонічними спазмами, зупинкою або регресом психомоторного розвитку та наявністю гіпсаритмії на ЕЕГ [3]. У 2009 р. вперше були проаналізовані генетичні причини епілептичних синдромів, що виникають протягом першого року життя, та запропоновані алгоритми їх діагностики [4].
Рання інфантильна епілептична енцефалопатія може проявлятися такими клінічними формами, як синдром Отахара (рання інфантильна епілептична енцефалопатія з патерном «спалах — пригнічення»), рання міоклонічна епілептична енцефалопатія (early myoclonic encephalopathy), інфантильні спазми, або синдром Веста (ISS), та синдром Леннокса — Гасо (LGS). Ці патологічні стани можуть бути викликані мутаціями в різних генах та мають схожу клінічну симптоматику. В останні роки ці синдроми класифікуються як епілептична енцефалопатія та енцефалопатія розвитку (developmental and epileptic encephalopathy), і хоча цей термін не входить до класифікації Міжнародної протиепілептичної ліги, ми вважаємо, що він найкраще демонструє спільність етології та клінічних проявів цих епілептичних синдромів раннього дитинства [5, 6]. Більше того, термін «енцефалопатія розвитку» є кроком уперед порівняно з «епілептичною енцефалопатією», оскільки при цих станах порушення розвитку в дітей є наслідком не лише епілептичних нападів, а й впливу генетичних мутацій [6].
Причинами розвитку ранніх епілептичних енцефалопатій можуть бути специфічні вади розвитку кори головного мозку, метаболічні порушення та десятки генетичних мутацій, що є безпосередньою причиною епілептичних нападів без супутніх метаболічних або структурних відхилень мозку. До таких мутацій належать ARX, CDKL5, SLC25A22, STXBP1 та SPTAN1 та багато інших.
На сьогодні виділяють 89 генетичних типів епілептичних енцефалопатій та енцефалопатій розвитку (developmental and epileptic encephalopathy) [7, 8]. У даній статті ми наводимо клінічну характеристику ранніх епілептичних енцефалопатій типу 1 (викликаних мутаціями гена ARX) і типу 2 (викликаних мутаціями гена СDKL5) та опис власного спостереження дитини з ранньою епілептичною енцефалопатією 2-го типу.
Рання інфантильна епілептична енцефалопатія типу 1
Причиною розвитку ранньої інфантильної епілептичної енцефалопатії типу 1 (EIEE1) є патогенні мутації гена ARX. EIEE1 є лише однією клінічною формою з широкого спектру розладів, викликаних порушенням даного гена. ARX — це перший ген, роль якого у розвитку ранньої епілептичної енцефалопатії доведена. Він пов’язаний з багатьма іншими порушеннями розвитку та вродженими аномаліями мозку. До ARX-залежних захворювань належать Х-зчеплена лісенцефалія з недорозвиненими геніталіями (OMIM 300215), синдром Proud (агенезія мозолистого тіла, аномальні геніталії, розумова відсталість) (OMIM 300004), інфантильні спазми без аномалій розвитку головного мозку (EIEE1, 308350), Х-зчеплена розумова відсталість, або синдром Partington (інтелектуальна недостатність і м’язова дистонія), синдромні та несиндромні форми розумової відсталості (300419), тяжка гідроцефалія, полімікрогірія з перивентрикулярною вузловою гетеротопією, неспецифічна Х-зчеплена інтелектуальна недостатність [8]. Хоча пацієнти чоловічої статі з мутаціями гена ARX зазвичай мають більш тяжкі порушення, жінки — носії мутації також можуть мати прояви захворювання [10].
Ген ARX (Aristaless-related homeobox) належить до родини гомеобоксних генів (гени, що детермінують процеси розвитку та диференціювання клітин) та відіграє критично важливу роль у ембріональному розвитку. Ген локалізований на короткому плечі Х-хромосоми у ділянці Xp22 і як фактор транскрипції є важливим для розвитку церебральних інтернейронів. Мутації у даному гені призводять до відсутності непірамідних та ГАМКергічних інтернейронів, зменшення вироблення білка ARX, що регулює активність інших генів, завдяки чому його називають фактором транскрипції. Ген ARX є одним із генів родини homeobox, що активуються під час раннього ембріонального розвитку та контролюють формування багатьох структур тіла. Зокрема, вважається, що білок ARX бере участь у розвитку головного мозку, підшлункової залози, яєчок, скелетних м’язів. У підшлунковій залозі, яєчках і скелетних м’язах білок ARX допомагає регулювати процес дозрівання клітин для виконання конкретних функцій (диференціювання). У мозку, що розвивається, білок ARX бере участь у міграції нейронів і формуванні зв’язків між нейронами. Зокрема, цей білок регулює гени, що відіграють певну роль у міграції спеціалізованих нейронів (інтернейронів) до їх належного місця розташування. Інтернейрони також передають сигнали між іншими нейронами [10].
EIEE1 успадковується за Х-зчепленим рецесивним типом. Ген ARX розташований на Х-хромосомі, що є однією з двох статевих хромосом. У пацієнтів чоловічої статі (в яких лише одна Х-хромосома) однієї зміненої копії гена в кожній клітині досить, щоб викликати захворювання. У жінок (в яких наявні дві Х-хромосоми) мутація зазвичай повинна відбуватися в обох копіях гена, щоб викликати захворювання. Однак у деяких випадках однієї зміненої копії гена ARX досить, оскільки хромосома X з нормальною копією гена ARX вимикається через процес, що називається X-інактивацією. На початку ембріонального розвитку в жінок одна з двох Х-хромосом інактивується в соматичних клітинах (окрім яйцеклітин і сперматозоїдів). Х-інактивація гарантує, що жінки, як і чоловіки, мають лише одну активну копію Х-хромосоми у кожній соматичній клітині тіла [8, 10, 11].
Мутації в гені ARX можуть становити до 5,2 % генетичної етіології ранньої епілептичної енцефалопатії та інфантильних спазмів у хлопчиків [12]. Найчастіше відзначається експансія першого чи другого поліаланінових трактів у гені. Тяжкість клінічних проявів зазвичай зростає зі збільшенням довжини експансій [10].
Клінічний фенотип EIEE1 схожий на інші типи ранньої епілептичної енцефалопатії з тяжкими порушенням нейророзвитку та характеризується раннім початком рефрактерної епілепсії (найпоширенішим типом нападу є інфантильні спазми), тяжкою затримкою розвитку, вираженими порушеннями тонусу та порушеним мовленнєвим і когнітивним розвитком. На ЕЕГ відзначається патерн «спалах — пригнічення» з трансформацією в гіпсаритмію [8].
Майже всі хворі діти мають порушення тонусу у вигляді дифузної гіпотонії або дистонії, у деяких випадках — з розвитком спастичності. Можуть відзначатися патологічні рухи у вигляді хореї або дистонії. Відзначені випадки синдрому пароксизмальної автономної нестабільності з дистонією (PAID), що проявляється тяжкою м’язовою дистонією та хореоатетозом кінцівок у поєднанні з лихоманкою, тахікардією, тахіпное та розширенням зіниць. Можуть відзначатися кіркові порушення зору та формування постнатальної мікроцефалії [8].
Для ЕІЕЕ1 характерні епілептичні напади у вигляді інфантильних спазмів, найчастіше у вигляді згинання шиї і тулуба та розгинання рук і ніг (спазми за типом «складного ножа»). Спазми тривають кілька секунд, об’єднуються у кластери тривалістю кілька хвилин. Найчастіше виникають після пробудження. Також можливе поєднання спазмів з тонічними, міоклонічними та парціальними нападами. Вік дебюту — до 1 року, припиняються до 5 років, але в багатьох дітей потім розвиваються інші типи судом, які повторюються протягом усього життя [9, 13].
На МРТ головного мозку можуть відмічатися дифузна атрофія або мальформації кори великих півкуль, гіпоплазія мозолистого тіла або хвостатих ядер, затримка мієлінізації білої речовини. Описано підвищення сигналу від блідих куль та огорожі, прогресуюча церебральна атрофія з вентрикуломегалією [8, 10].
На сьогодні ефективне лікування ЕІЕЕ1 відсутнє. Протиепілептичні препарати, зокрема бензодіазепіни, вальпроати, леветірацетам, зонісамід і фенобарбітал, мають обмежену ефективність. Описані окремі випадки ефективного застосування піридоксину (вітаміну В6). Кетогенна дієта або дієта Аткінса можуть бути корисними в деяких пацієнтів. Найбільш ефективними терапевтичними методами є застосування вігабатрину в поєднанні з кортикостероїдними гормонами (адренокортикотропний гормон (АКТГ), преднізолон, метилпреднізолон). Альтернативою можуть бути нові протиепілептичні препарати, зокрема фенфлурамін, ганаксолон, каннабідіол. Баклофен і клоназепам застосовують для лікування тонусних порушень [8, 14].
Рання інфантильна епілептична енцефалопатія типу 2
Рання інфантильна епілептична енцефалопатія типу 2 (EIEE2) викликається мутаціями гена CDKL5, який кодує циклінзалежну протеїнкіназу 5-го типу (cyclin-dependent kinase-like 5). Патогенні мутації даного гена можуть включати делеції, трункації, сплайс-варіанти та міссенс-мутації. Білок CDKL5 широко поданий у нейронах мозку і відіграє важливу роль у регуляції проліферації клітин, нейрональній міграції, рості аксонів, морфогенезі дендритів і розвитку синапсів [15].
Ген був вперше картований у ділянці хромосоми Xp22.3 M. Montini та співавт. у 1998 р., проте ідентифікація мутацій у даному гені була проведена у 2004 р. Тип успадкування ЕІЕЕ2 — X-зчеплений домінантний [16, 17].
Мутації в даному гені переважно відзначаються в дівчаток з раннім початком рефрактерних епілептичних нападів, тяжкою загальною затримкою розвитку, відсутністю або тяжким розладом мовлення, стереотипними рухами та (в деяких випадках) затримкою росту окружності голови. У деяких випадках клінічна картина нагадує синдром Ретта, що раніше давало підставу діагностувати в дівчаток з мутаціями гена CDKL5 атиповий синдром Ретта. L.S. Weaving та співавт. у 2004 р. запропонували називати даний синдром атиповим синдромом Ретта. Однак, згідно з сучасною класифікацією, в каталозі ОМІМ даний синдром визначений як рання епілептична енцефалопатія 2-го типу. Більш сучасна назва захворювань, пов’язаних з мутаціями гена CDKL5, — «розлади дефіциту CDKL5» (CDKL5-deficiency disorder) [8, 16].
Встановлено, що білок — продукт гена CDKL5 — є ферментом, що бере участь у фосфорилюванні білкового продукту гена МЕСР2, відповідального за розвиток синдрому Ретта, тому клінічна картина ЕІЕЕ2 нагадує синдром Ретта [17].
У дослідженнях мутації гена CDKL5 відзначаються у 8 % жінок з раннім початком епілептичних нападів (до 9-го місяця життя) та у 28 % дівчаток з інфантильними спазмами. CDKL5-залежна епілептична енцефалопатія зустрічається приблизно в 1 з 40 000–60 000 новонароджених. У хлопчиків мутації цього гена виявляються рідше, ніж у дівчаток, однак переважно викликають більш тяжкі прояви з раннім початком тонічних або міоклонічних нападів, резистентних інфантильних спазмів, тяжкою загальною затримкою розвитку, кортикальним порушенням зору, порушеннями тонусу, розладами сну та стереотипними рухами кінцівок в деяких пацієнтів. У більшості уражених дітей формується вторинна мікроцефалія. Вираженість інтелектуальних порушень може значно відрізнятися в різних пацієнтів. Можливий розвиток лише когнітивних порушень без виникнення епілепсії або структурних змін у мозку. Деякі діти мають тяжкі проблеми з харчуванням, зокрема гастроезофагеальний рефлюкс та порушення ковтання, та можуть потребувати встановлення гастростоми [8, 16–18].
Найчастіше у дітей з ЕІЕЕ2 відзначаються кластерні інфантильні спазми, що можуть починатися у віці 1 міс. та супроводжуватися розвитком гіпсаритмії на ЕЕГ. Середній вік початку нападів — 6 тижнів життя, але в 90 % дітей напади дебютують до 3-місячного віку. Можливі також тонічні напруження кінцівок з девіацією очей, атонічні та міоклонічні напади. У клінічному перебігу ЕІЕЕ2 виділяють 3 стадії [15]:
1) ранній початок рефрактерних епілептичних нападів;
2) розвиток епілептичної енцефалопатії;
3) трансформація у рефрактерну мультифокальну або міоклонічну епілепсію.
Рухові порушення при ЕІЕЕ2 можуть проявлятися стереотипними рухами верхніх кінцівок та голови, дистонічними рухами руками, неможливістю брати предмети в руки та в рідкісних випадках — розвитком spasmus nutans (асиметричний маятникоподібний ністагм, кивання головою та тортиколіз) [8].
МРТ головного мозку може виявляти прогресуючу церебральну та церебелярну атрофію з розширенням субарахноїдальних просторів, борозен, вентрикуломегалією та стоншенням білої речовини півкуль і мозолистого тіла [8].
Лікування ЕІЕЕ2 на сьогодні остаточно не розроблене. Напади в більшості випадків є рефрактерними до терапії. Препаратами вибору є вігабатрин та АКТГ. Резервними препаратами — фенобарбітал і карбамазепін. Кетогенна дієта може мати частковий ефект, проте напади зазвичай залишаються рефрактерними.
Сподівання у лікуванні ЕІЕЕ2 пов’язані з таргетною терапією. Нещодавно завершено 3-тю фазу клінічного дослідження MARIGOLD, що вивчало ефективність використання перорального ганаксолону в дітей та осіб молодого віку з розладами дефіциту CDKL5. ГАМКергічна дисфункція, про яку повідомляється у пацієнтів із CDKL5, обґрунтовує застосування ганаксолону. Повідомляють також, що інші нейростероїди, особливо прегненолон, здатні запобігати морфологічним дефектам у нейронах, позбавлених білка CDKL5. Одним із метаболітів прегненолону є алопрегнанолон, який за своєю структурою та механікою схожий на ганаксолон [19].
Дослідницькою групою на чолі з професором O. Dulac показана ефективність комбінованої терапії з застосуванням вігабатрину та зонісаміду в лікуванні інфантильних спазмів при ЕІЕЕ2 [20].
Як ілюстрацію надаємо клінічне спостереження дівчинки з ранньою епілептичною енцефалопатією 2-го типу, що є нашим власним досвідом встановлення діагнозу та ведення пацієнта з встановленою мутацією у гені СDKL5.
Дівчинка П., 1 рік 7 міс. Батьки вперше звернулися до відділення психоневрології нашого інституту з 4 міс. зі скаргами на рефрактерні епілептичні напади, регрес статокінетичного та психоемоційного розвитку.
З анамнезу відомо, що дитина народилася від 1-ї вагітності, 1-х пологів. Вагітність перебігала на фоні раннього гестозу, гострої респіраторної вірусної інфекції у 1-й та 2-й половині вагітності, лабіального герпесу на 7-му міс. Пологи шляхом екстреного кесаревого розтину у зв’язку зі слабкістю пологової діяльності (перейми тривали 19 годин), зниженням серцебиття плода. Народилася з масою тіла 3550 г, зріст 50 см, окружність голови 35 см, окружність грудної клітки 36 см. За шкалою Апгар отримала 6/7 балів. Закричала відразу. Виписана додому на 5-ту добу у задовільному стані. Утримувала голову з 2 міс., переверталася зі спини та живота з 9 міс. Стояла з підтримкою з 13 міс., самостійно стояла біля опори з 18 міс. Окремі склади вимовляла з 2 р. Регрес у розвитку відмічався в 4 та 7 міс.
Дівчинка захворіла у віці 3 міс., коли в неї у стані неспання спостерігалися підкочування очних яблук угору та їх посмикування протягом кількох секунд. Наступного дня виникли серійні напади у вигляді кивків головою з розведенням верхніх кінцівок у сторони, по 5–7 нападів у серії, до 10 нападів на добу. За місцем проживання призначені діакарб з аспаркамом, магне В6, пантогам, які дитина приймала протягом 1 міс. За цей час кількість нападів зросла до 50 серій на день, по 20 елементів у серії.
З 4 міс. відмічався регрес у розвитку: дитина перестала посміхатися, гулити, фіксувати погляд, перевертатися. У цьому віці на ЕЕГ вперше виявлена гіпсаритмія, у зв’язку з чим призначений вальпроат натрію (депакін), на фоні якого напади стали менш частими. Через кілька тижнів до терапії доданий вігабатрин, на фоні прийому якого частота нападів зменшилась на 50 %. У подальшому кількаразово проходила курси лікування у відділенні психоневрології ДУ «ІПАГ імені акад. О.М. Лук’янової НАМН». Отримувала курси прийому дексаметазону, на фоні якого з’явились ознаки екзогенного гіперкортицизму, була неспокійною, постійно плакала. У віці 7 міс. знову відбувся регрес розвитку — перестала перевертатися, фіксувати погляд, цікавитися іграшками, водночас на ЕЕГ була повторно зафіксована гіпсаритмія. В подальшому навички відновилися у віці 9 міс.
Через 2–3 міс. після відміни дексаметазону відновилися напади у вигляді підведення очей угору з затримкою дихання. Напади виникали під час годування, провокувалися піднесенням ложки до рота. За добу мати відмічала близько 100 одиничних нападів. Продовжувала отримувати вальпроат натрію, вігабатрин. До терапії був доданий метіпред 4 мг 3 р/д.
У подальшому неодноразово проводилася корекція терапії — дитина отримувала леветирацетам (кеппру), клобазам (фрізіум), етосукцимід, клоназепам, вігабатрин, преднізолон. Після курсу АКТГ (сінактен-депо) нападів не було більше ніж 1 міс., потім стався рецидив.
На фоні введення топірамату (топамакс) 25 мг 2 р/д відзначалась позитивна динаміка — зменшилась частота епізодів підведення очей угору до 3–4 р/день, через деякий час з’явились «світлі дні», коли напади були відсутніми.
Неврологічний статус (у віці 1 р. 6 міс.). Окружність голови 45 см (мікроцефалія). Задовільного харчування. Шкірні покриви чисті. Соматичний статус без відхилень. Поведінка гіперактивна. На огляд реагує спокійно. Вимовляє окремі склади. Реагує на своє ім’я. Розуміє звернення на елементарному рівні. Відсутній вказівний жест. Інструкції практично не виконує. Функції черепних нервів не порушені. Не жує. Об’єм рухів у верхніх і нижніх кінцівках не порушений. Порушення тонусу відсутні. Самостійно не сідає, не повзає, не ходить. Стоїть біля опори кілька хвилин. Крокує при підтримці на рівних ногах. Сидить пасивно посаджена нетривало, з рівною спиною. Черевні рефлекси живі, симетричні. Сухожилкові рефлекси живі, D = S. Симптом Бабінського двобічний.
/93.jpg)
ЕЕГ у віці 1 рік: ознаки епілептиформної активності у вигляді комплексів «спайк-хвиля» у лівих центрально-скроневих відведеннях, періодично у вигляді генералізованих комплексів «спайк — поліспайк — повільна хвиля» в поєднанні з високоамплітудними загостреними повільними хвилями з амплітудним акцентом у лівих центрально-скроневих ділянках.
ЕЕГ-моніторинг нічного сну (1 р. 7 міс.): епілептиформна активність реєструється під час сну в вигляді поодиноких гострих хвиль, спайків у лівій скроневій ділянці.
Генетичний аналіз крові методом секвенування нової генерації (NGS) і тестування на делеції/дуплікації 187 генів (лабораторія Invitae, м. Сан-Франциско, США) виявив гомозиготну патогенну мутацію c.3083C>T (p.Thr1028Met) у 21-му екзоні гена СDKL5. Даний варіант гена полягає у заміні треоніну метіоніном у кодоні 1028 білка CDKL5 (p.Thr1028Met). Цей варіант наявний у базах даних мутацій (rs762576315, ExAC 0,01 %), однак раніше не описаний у пацієнтів з СDKL5-залежними захворюваннями.
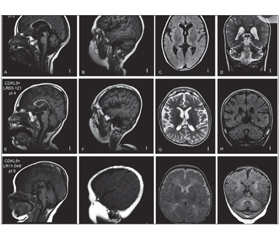
МРТ головного мозку 1,5 Тл (1 рік) — ознак вогнищевої патології не виявлено. Локальна зовнішня фронтотемпоральна гідроцефалія. Mega cysterna magna.
МРТ головного мозку (3 Тл) — бітемпоральне розширення зовнішніх лікворних просторів (bening external hydrocephaly). Неспецифічна гіперінтенсивність білої речовини скроневих ділянок. Даних щодо мезіального склерозу, ФКД не отримано.
Висновки
У клінічній практиці ми — дитячі неврологи — звикли користуватися терміном «епілептична енцефалопатія». Проте останнім часом набув поширення термін «епілептична енцефалопатія та енцефалопатія розвитку» (developmental and epileptic encephalopathy). На нашу думку, він точніше відображає патологічні стани, при яких мутації у критично важливих для розвитку нервової системи генах призводять до розвитку резистентних епілептичних нападів у дітей раннього віку та як наслідок — до порушень когнітивного, моторного та мовленнєвого розвитку.
Епілептичні енцефалопатії й енцефалопатії розвитку є групою тяжких розладів, що характеризуються затримкою або регресом когнітивних і поведінкових навичок, які розвиваються внаслідок резистентних епілептичних нападів. З огляду на клінічну та генетичну гетерогенність цих розладів, секвенування нової генерації є важливою складовою стратегії обстеження пацієнтів з метою ідентифікації генетичної етіології розладу та призначення таргетної протиепілептичної терапії.
Ранні епілептичні енцефалопатії 1-го та 2-го типів є рідкісними генетичними варіантами моногенних епілептичних енцефалопатій з Х-зчепленим типом успадкування, що призводять до розгорнутої клінічної картини ураження не лише шляхом провокації епілептичних нападів, а й завдяки впливу генетичних мутацій на різні ланки диференціювання, міграції нейронів і формування міжнейронних зв’язків. Якнайшвидше виявлення характерних типів нападів, специфічних патернів на ЕЕГ і проведення генетичної діагностики допомагають підібрати максимально вдалу комбінацію препаратів для запобігання прогресуванню епілептичних нападів і розвитку тяжкої когнітивної недостатності.
Важливим результатом даної роботи є ідентифікація мутації c.3083C>T (p.Thr1028Met) гена СDKL5, раніше не описаної в дітей з СDKL5-залежними захворюваннями.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 14.05.2021
Рецензовано/Revised 28.05.2021
Прийнято до друку/Accepted 05.06.2021
Список литературы
1. Ohtahara S., Ishida T., Oka E., Yamatogy Y., Inoue H. On the specific age-dependent epileptic syndromes: The early-infantile epileptic encephalopathy with suppression-burst. No To Hattatsu. 1976. № 8. Р. 270-280.
2. Berg A.T., Berkovic S.F., Brodie M.J., Buchhalter J., Cross J.H., van Emde Boas W., Engel J., Glauser T.A., Mat-hern G.W., Moshe S.L., Nordli D., Plouin P., Scheffer I.E. Revised terminology and concepts for organization of seizures and epilepsy: Report of the ILAE Commission on Classification and Terminology. Epilepsia. 2010. № 51. Р. 676-685.
3. Stafstrom C.E., Kossoff E.M. Epileptic Encephalopathy in Infants and Children. Epilepsy Curr. 2016. № 16(4). Р. 273-279. doi: 10.5698/1535-7511-16.4.273.
4. Deprez L., Jansen A., DeJonghe P. Genetics of epilepsy syndrome starting in the first year of life. Neurology. 2009. № 72. Р. 273-281. [PubMed: 19153375, relatedcitations]. [FullText].
5. Nieh S.E., Sherr E.H. Epileptic encephalopathies: new genes and new pathways. Neurotherapeutics. 2014. № 11. Р. 796-806.
6. Lopez-Santiago L., Isom L.L. Dravet Syndrome: A Developmental and Epileptic Encephalopathy. Epilepsy Curr. 2019. № 19. Р. 51-53. doi: 10.1177/1535759718822038.
7. Specchio N., Curatolo P. Developmental and epileptic encephalopathies: what we do and do not know. Brain. 2021 January. Vol. 144. Issue 1. P. 32-43. https://doi.org/10.1093/brain/awaa371
8. Mirzaa G.M., Paciorkowski A.R., Marsh E.D. et al. CDKL5 and ARX mutations in males with early-onset epilepsy [published correction appears. Pediatr. Neurol. 2013 Jul. № 49(1). 74. Alkhateeb, Asem [added]]; Pediatr. Neurol. 2013. № 48(5). Р. 367-377. doi: 10.1016/j.pediatrneurol.2012.12.030.
9. Shoubridge C., Fullston T., Gécz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum. Mutat. 2010 Aug. № 31(8). Р. 889-900.
10. Kato M., Saitoh S., Kamei A., Shiraishi H., Ueda Y., Akasaka M., Tohyama J., Akasaka N., Hayasaka K. A Longer Polyalanine Expansion Mutationin the ARX Gene Causes Early Infantile Epileptic Encephalopathy with Suppression-Burst Pattern (Ohtahara Syndrome). The American Journal of Human Genetics. 2007. Vol. 81. Issue 2. P. 361-366. ISSN 0002-9297. https://doi.org/10.1086/518903. https://www.sciencedirect.com/science/article/pii/S0002929707612002
11. Mattiske T., Tan M.H., Dearsley O. et al. Regulating transcriptional activity by phosphorylation: A new mechanism for the ARX homeodomain transcription factor. PLoS One. 2018. № 13(11). e0206914. Published 2018 Nov 12. doi: 10.1371/journal.pone.0206914.
12. Kato M., Das S., Petras K. et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 2004. № 23(2). Р. 147-159. doi: 10.1002/humu.10310.
13. Takeshita Y., Ohto T., Enokizono T. et al. Novel ARX mutation identified in infantile spasm syndrome patient. Hum. Genome Var. 2020. Vol. 7. № 9. https://doi.org/10.1038/s41439-020-0094-2
14. Shbarou R. Current Treatment Options for Early-Onset Pediatric Epileptic Encephalopathies. Curr. Treat. Options Neurol. 2016. Vol. 18. № 44. https://doi.org/10.1007/s11940-016-0428-z
15. Olson H.E., Demarest S.T., Pestana-Knight E.M. et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019. № 97. Р. 18-25. doi: 10.1016/j.pediatrneurol.2019.02.015.
16. Bahi-Buisson N., Bienvenu T. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Mol. Syndromol. 2012. № 2(3–5). Р. 137-152. doi: 10.1159/000331333.
17. Дадали Е.Л., Акимова И.А., Коновалов Ф.А. и др. Клинико-генетические особенности пациентов с ранней эпилептической энцефалопатией 2-го типа, обусловленной мутациями в гене CDKL5. Русский журнал детской неврологии. 2019. № 14(3). С. 28-36.
18. Szafranski P., Golla S., Jin W. et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur. J. Hum. Genet. 2014, 15 October.
19. Vaitkevicius H., Husain A.M., Swisher C.B. et al. Phase 2 open-label study of intravenous ganaxolone for the treatment of refractory status epilepticus. Presented at: Neurocritical Care Society 17th Annual Meeting. 2019 October 15–18. Vancouver, British Columbia. Oral Abstract. 2.
20. Melikishvili G., Epitashvili N., Tabatadze N. et al. New insights in phenomenology and treatment of epilepsy in CDKL5 encephalopathy. Epilepsy Behav. 2019. № 94. Р. 308-311. doi: 10.1016/j.yebeh.2019.02.013.
21. Евтушенко С.К. Разрушительные и труднокурабельные формы эпилепсии и эпилептические энцефалопатии у детей. Международный неврологический журнал. 2012. № 6(52). С. 15-26.

/91.jpg)
/92.jpg)
/93.jpg)
/94.jpg)