Вступ
Прогресуючі м’язові дистрофії є клінічно, генетично і біохімічно гетерогенною групою захворювань незапального характеру, в основі яких лежить первинний дефект м’язових волокон, що супроводжується характерними, але часто непатогномонічними патологічними ознаками [1]. Клінічно вони характеризуються прогресуючою м’язовою слабкістю, атрофією м’язів і руховими порушеннями.
Статистика
Як окремі розлади м’язові дистрофії зустрічаються відносно рідко, але в цілому вони становлять значну частку пацієнтів з нервово-м’язовими захворюваннями. Загальна поширеність всіх м’язових дистрофій — 16,14 на 100 000 [2].
У дорослому віці міотонічна дистрофія є найбільш поширеною формою, зачіпаючи приблизно 10,6 на 100 000 чоловік, а лице-лопатково-плечова м’язова дистрофія зустрічається приблизно в 3,0 на 100 000 чоловік [3]. З кінцівково-поясної м’язової дистрофії (КПМД) рецесивні форми зустрічаються частіше, ніж домінуючі варіанти. Поширеність усіх підтипів КПМД коливається від 4 до 7 на 100 000, залежно від географічного та етнічного походження. Більша частина рецесивних випадків КПМД обумовлена кальпаїнопатіями, дисферлінопатіями і саркогліканопатіями [4, 5]. Поширеність вроджених м’язових дистрофій значно варіюється залежно від регіону. Точкова поширеність коливається від 0,68 до 2,5 на 100 000 чоловік [6, 7].
Лице-лопатково-плечова м’язова дистрофія (FSHD, Ландузі — Дежерина) є м’язовою дистрофією, що виникає як у дітей, так і дорослих пацієнтів. Жінки хворіють в три рази частіше за чоловіків. Mah і співавт. повідомляють про загальну поширеність у всіх вікових групах 3,95 на 100 000 [2]. Norwood по північній частині Великої Британії повідомляє, що FSHD є другою за поширеністю формою прогресуючих м’язових дистрофій, як і дистрофінопатії [6]. У цілому по Європі показник поширеності FSHD становить приблизно 1 на 10 000 [8].
До числа прогресуючих міодистрофій відноситься й окулофарингеальна м’язова дистрофія, яка зустрічається в двох генетичних варіантах: автосомно-рецесивному та автосомно-домінантному. Поширеність по всьому світу — від 0,5 до 1 на 100 000 населення [9].
Автосомно-домінантна дистрофічна міотонія 1-го типу (ДМ1) і дистрофічна міотонія 2-го типу (ДМ2, проксимальна міотонічна міопатія), на відміну від недистрофічної міотонії, є прогресуючими мультисистемними захворюваннями, що характеризуються клінічною тріадою: прогресуючою слабкістю, міотонією і катарактою з раннім початком. Поширеність дистрофічної міотонії в різних популяціях сильно варіюється і різниться між ДМ1 і ДМ2. Theadom і співавт. показали діапазон поширеності ДМ від 0,5 до 18,1 на 100 000 чоловік [10], а метааналіз, проведений Mah і співавт., довів, що поширеність ДМ становила 8,26 на 100 000 [2]. Серед європейців поширеність дистрофічної міотонії оцінена в 12,5 на 100 000 [11].
Статистичні дані щодо поширеності м’язової дистрофії в Україні недостатні. Є окремі дані щодо спінальної м’язової дистрофії (СМА) — 271 дитина, м’язової дистрофії Дюшена — 358 дітей (МОЗ України). Також, за даними Донецького обласного клінічного центру нейрореабілітації за період 2001–2012 рр., зареєстровано 152 дитини з первинним м’язовим захворюванням, 107 — із СМА та 89 дітей — із спадковою мотосенсорною поліневропатією [12].
Методи
Безумовно, важлива роль у діагностиці м’язових дистрофій належить збору анамнезу та клінічному обстеженню пацієнтів. Разом з тим поєднання клінічних ознак і аналіз можливого типу успадкування дозволяють запідозрити конкретні форми м’язової дистрофії та напрямки подальшого дослідження, хоча збіги між генетично відмінними формами ускладнюють діагностичний пошук. Клініцистам все більшою мірою необхідно спиратися на дані патогістологічних і електрофізіологічних досліджень, а також на дані генетики та візуалізаційних методів для більш точної диференціальної діагностики.
Електроміографія (ЕМГ) є значущим методом ранньої діагностики м’язових дистрофій, у тому числі і диференціальної діагностики нервово-м’язової патології, визначення стадії і активності міодистрофічного процесу. При ЕМГ, як правило, виявляються зниження амплітуди потенціалів при достатній їх частоті, а також вкорочення і поліфазний характер потенціалів дії рухових одиниць [1, 3, 13]. Також при деяких формах м’язової дистрофії за даними ЕМГ спостерігаються нейрогенні ознаки (висока амплітуда, поліфазного характеру), які відображають гіпертрофію і спостерігаються частіше при повільно прогресуючих процесах [14, 15]. Крім зазначених змін, при окремих формах міодистрофії спостерігається патологічна спонтанна активність з міотонічними розрядами в окремих м’язах [16]. Таким чином, ЕМГ є перевіреним, добре вивченим і визнаним методом, що дозволяє визначати функціональний стан м’яза, наявність або відсутність поточного патологічного процесу в м’язі, у тому числі наявність первинно-м’язового ураження, однак зміни не мають високої специфічності [17].
Лабораторні методи дослідження, включаючи біохімічний аналіз крові, використовуються при проведенні диференціальної діагностики м’язових дистрофій з іншими нервово-м’язовими захворюваннями, включаючи мітохондріальну, автоімунну, а також ендокринну патологію [18]. Біохімічні методи дослідження дозволяють виявити підвищення активності м’язових ферментів в сироватці, найбільш специфічного — креатинфосфокінази (КФК). Пацієнти з ознаками прогресуючої м’язової слабкості повинні пройти біохімічний скринінг з визначенням показників, що дозволяє виявити показники рабдоміолізу, в тому числі і для проведення диференціальної діагностики м’язових дистрофій з іншими нервово-м’язовими захворюваннями.
Ультразвук (УЗД) — добре розроблений і перевірений метод діагностичної візуалізації при оцінці пацієнтів з підозрою на м’язові розлади [19]. Основним недоліком УЗД є те, що його застосування обмежене поверхневими групами м’язів. Цей ефект стає ще більш вираженим, коли кілька груп м’язів перекриваються [20].
За даними англомовної літератури, використання магнітно-резонансної томографії (МРТ) для діагностики, оцінки прогресування м’язової дистрофії та класифікації підтипів отримало велике поширення в останні десятиліття. МРТ м’язів дає нам уявлення щодо багатьох аспектів будови і функції скелетних м’язів. Описані конкретні м’язи, які беруть участь в цих м’язових захворюваннях. Такі специфічні патерни вселяють надію на те, що розподіл змін у м’язах може допомогти в діагностиці захворювання [21].
З огляду на те, що МРТ — неінвазивна методика, її можна багаторазово повторювати, це значно поліпшує якість досліджень при захворюваннях. Використання МРТ для вивчення м’язової дистрофії та виявлення специфічних патернів м’язового залучення може сформулювати діагностичні напрямки, значно скоротити діагностичний пошук. Вагома роль МРТ м’язів в оцінці генетичних захворювань м’язів [22]. Число генів, які викликають м’язову дистрофію, продовжує збільшуватися, і доступність генетичного тестування для цих станів створила потребу в поліпшенні фенотипування цих розладів, щоб оптимізувати вибір генетичних тестів і досліджувати варіанти генів невизначеної значущості [23]. МРТ допомагає клініцисту направляти генетичне тестування у пацієнтів з результатами, що частково збігаються, або неспецифічними клінічними фенотипами.
Поєднання клініко-лабораторних та інструментальних методів, таких як МРТ, може скорочувати діагностичний пошук [21]. Хоча секвенування наступного покоління (NGS) надає безліч генетичних даних, для інтерпретації часто потрібне глибоке фенотипування, включаючи клінічні, лабораторні та візуальні дані.
В цілому МРТ м’язів має більш високу діагностичну цінність при повільно прогресуючих захворюваннях, при яких вибіркові патерни м’язового залучення можуть бути виявлені протягом тривалого періоду часу [24]. МРТ м’язів можна використовувати як діагностичний інструмент для опису ступеня і характеру ураження м’язів і для визначення локалізації майбутньої біопсії м’язів при необхідності [25–27].
Останніми роками спостерігається стійкий прогрес у виявленні генетичних захворювань м’язів, а також в зусиллях з розробки лікування цих захворювань. Отже, необхідні чутливі й об’єктивні нові методи для виявлення та моніторингу м’язової патології. Магнітно-резонансна томографія пропонує безліч потенційних біомаркерів тяжкості захворювання при м’язовій дистрофії. МРТ м’язів допомагає не тільки у ранній діагностиці захворювання та подальшому спостереженні за його прогресуванням, але й в оцінці ефективності лікування.
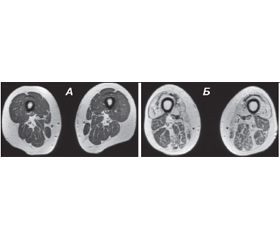
МРТ — метод, заснований на отриманні серій зображень у фронтальній і аксіальній площині зрізів з використанням певних імпульсних послідовностей: Т1-ЗЗ, Т2-ЗЗ, STIR. Ці послідовності допомагають відрізнити м’язову тканину від жирової, набряк і жирову дегенерацію. Особливо важливо застосування правильних послідовностей у випадках, коли є підозри на запальні зміни в м’язовій тканині. Застосування рейтингових шкал візуальної оцінки дистрофічних змін м’язової тканини (Mercuri et al., 2002; Kornblum et al., 2006; Fischer et al., 2008; Lamminen А., 1990; модифікація Jungbluth Н. et al., 2004) дозволяє оцінити ступінь дегенерації уражених м’язів (рис. 1).
/20.jpg)
Нами виконано більш ніж 50 обстежень м’язів пацієнтам як із первинно-м’язовою патологією, так і з нервово-м’язовим типом ураження. На основі нашого досвіду ми розробили протокол МРТ-обстеження пацієнтів із ураженням м’язів, який дозволяє за нетривалий час обстеження отримати максимум інформації для подальшої діагностики. Застосування МРТ м’язів допомогло нам як виявити у наших пацієнтів генетичні форми ураження м’язів, так й діагностувати різні форми вторинних міопатій, зокрема запальні, ендокринні, стероїдні та, в тому числі, хвороби накопичення із залученням м’язів.
Висновки
Прогресуючі м’язові дистрофії відносяться до тяжкокурабельної патології, яка, на жаль, дуже часто пізно діагностується. Тому при перших підозрах на наявність у пацієнта м’язової патології доцільно проводити діагностичні заходи, які включають загальні клінічні обстеження, ЕМГ, генетичне тестування та застосування МРТ м’язів, що дають підставу для призначення терапії.
На жаль, на сьогодні не існує патогенетичної терапії для більшості м’язових дистрофій, а традиційні методи лікування, що ґрунтуються на імунодепресантах, таких як кортикостероїди або протизапальні препарати, спрямовані лише на пом’якшення симптомів. Водночас в останні кілька років з’явились препарати для лікування м’язової дистрофії Дюшена (МДД), спінальної м’язової атрофії (СМА), а також деяких хвороб накопичення з ураженням м’язів (наприклад, хвороба Помпе). Такі препарати, як аталурен для лікування МДД, нусінерсен та рисдиплам для лікування СМА, вже зареєстровані в Україні.
З огляду на високу вартість цих препаратів виникає необхідність розробки ефективного алгоритму верифікації форм м’язових дистрофій, що є дуже важливим не тільки для підтвердження діагнозу в пацієнтів, а і для призначення патогенетичної терапії. Також виникає необхідність контролю за ефективністю лікування. МРТ м’язів може стати як об’єктивним маркером оцінки патогенетичного лікування, так і маркером оцінки прогресування м’язових дистрофій.
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 02.10.2021
Рецензовано/Revised 21.10.2021
Прийнято до друку/Accepted 28.10.2021
Список литературы
1. Emery A. The muscular dystrophies. Lancet. 2002. 359. 687-95. doi: 10.1016/S0140-6736(02)07815-7.
2. Mah J., Korngut L., Fiest K. et al. A systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can. J. Neurol. Sci. 2016. 43(1). 163-177. doi: 10.1017/cjn.2015.311.
3. Mercuri E., Muntoni F. Muscular dystrophies. Lancet. 2013. 381 (9869). 845-60. doi: 10.1016/S0140-6736(12)61897-2.
4. Magri F., Nigro V., Angelini C. et al. The Italian LGMD registry: Relative frequency, clinical features, and differential diagnosis: LGMD Italian registry. Muscle & Nerve. 2017. 55. 55-68. doi: 10.1002/mus.25192.
5. Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract. Neurol. 2009. 9(6). 314-23. doi: 10.1136/jnnp.2009.193938.
6. Norwood F., Harling C., Chinnery P. et al. Prevalence of gene-tic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009. 132. 3175-3186. doi: 10.1093/brain/awp236.
7. Darin N., Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul. Disord. 2000. 10. 1-9. doi: 10.1016/s0960-8966(99)00055-3.
8. Sacconi S., Salviati L., Desnuelle C. Facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta. 2014. 1852(4). 607-14. doi: org/10.1016/j.bbadis.2014.05.021.
9. Marsh E., Robinson D. A case of rare recessive oculopharyngeal muscular dystrophy (OPMD) coexisting with hereditary neuropathy with liability to pressure palsies (HNPP). Clin. Neurol. Neurosurg. 2008. 110. 525-8. doi: 10.1016/j.clineuro.2008.02.007.
10. Theadom A., Rodrigues M., Roxburgh R., Balalla S., Higgins C., Bhattacharjee R., Jones K., Krishnamurthi R., Feigin V. Prevalence of Muscular Dystrophies: A Systematic Literature Review. Neuroepidemiology. 2014. 43. 259-268. doi: 10.1159/000369343. doi.org/10.1159/000369343.
11. Thornton C. Myotonic dystrophy. Neurol. Clin. 2014. 32(3). 705. doi: 10.1016/j.ncl.2014.04.011.
12. Евтушенко С., Шаймурзин М., Евтушенко О., Евту-шенко И. Нейромышечные заболевания у детей: монография. Донецк: Изд-во «Ноулидж» (донецкое отделение), 2014. 218 с.
13. Katharine M. Bushby and Louise V. Anderson. Methods in Molecular Medicine. Muscular Dystrophy: Methods and Protocols. Ed. by K.M.D. Bushby, L.V.B. Anderson.
14. Rowinska-Marcinska K., Szmidt-Salkowska E., Fidzianska A. et al. Atypical motor unit potentials in Emery-Dreifuss muscular dystrophy (EDMD). Clin. Neurophysiol. 2005. 116(11). 2520-2527. doi: 10.1016/j.clinph.2005.01.017.
15. Zivari Adab H., Firoozabadi S.M., Chalavi S., Maghooli K. Simulation and analysis of needle electromyogram in Emery-Dreifuss muscular dystrophy by using line source model. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2008. 2008. 338-342. doi: 10.1109/IEMBS.2008.4649159.
16. Petty R., Thomas P., Landon D. Emery-Dreifuss syndrome. J. Neurol. 1986. 233(2). 108-114. doi: 10.1007/BF00313856.
17. Hehir M., Logigian E. Electrodiagnosis of myotonic disorders. Phys. Med. Rehabil. Clin. N. Am. 2013. 24(1). 209-220. doi: org/10.1016/j.pmr.2012.08.015.
18. Евтушенко С., Морозова Т., Шестова Е. с соавт. Синдром мышечной гипотонии у новорожденных и детей раннего возраста. Донецк, 2008. 240 с.
19. Pillen S., Arts I.M., Zwarts M.J. Muscle ultrasound in neuromuscular disorders. Muscle Nerve. 2008. 37. 679-693. doi: 10.1002/mus.21015.
20. Pillen S., Keimpema M., Bievelstein R., Verrips A., Kruijsbergen-Raijmann W., Zwarts M. Skeletal muscle ultrasonography: visual versus quantitative evaluation. Ultrasound Med. Biol. 2006. 32. 1315-1321. doi: 10.1016/j.ultrasmedbio.2006.05.028.
21. Mercuri E., Pichiecchio A., Allsop J., Messina S., Pane M., Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J. Magn. Reson. Imaging. 2007. 25(02). 433-440. doi: 10.1002/jmri.20804.
22. Leung D. Magnetic resonance imaging patterns of muscle involvement in genetic muscle diseases: a systematic review. J. Neurol. 2017. 264. 1320-1333. doi: 10.1007/s00415-016-8350-6.
23. Straub V., Murphy A., Udd B. 229th ENMC international workshop: limb girdle muscular dystrophies-nomenclature and reformed classification, Naarden, the Netherlands, 17-19 March 2017. Neuromuscul. Disord. 2018. 28. 702-710. doi: 10.1016/j.nmd.2018.05.007.
24. Jungbluth H. Myopathology in times of modern imaging. Neuropathol. Appl. Neurobiol. 2017. 43(01). 24-43. doi: 10.1111/nan.12385.
25. Wattjes M., Kley R., Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur. Radiol. 2010. 20. 2447-2460. doi: 10.1007/s00330-010-1799-2.
26. Ten Dam L., van der Kooi A., Verhamme C., Wattjes M.P., de Visser M. Muscle imaging in inherited and acquired muscle diseases. Eur. J. Neurol. 2016. 23. 688-703. doi: 10.1111/ene.12984.
27. Quijano-Roy S., Carlier R., Fischer D. Muscle imaging in congenital myopathies. Semin. Pediatr. Neurol. 2011. 18. 221-229. doi: 10.1016/j.spen.2011.10.003.

/20.jpg)