Концепція моноклональної гаммапатії ренального значення (МГРЗ) була запропонована Міжнародною групою з вивчення ураження нирок і моноклональної гаммапатії (International Kidney and Monoclonal Gammopathy Research group) [1] та відображає патологічний стан, обумовлений проліферацією клону В-клітин або плазматичних клітин, що не досягає критеріїв, необхідних для початку лікування за онкогематологічними показаннями, але продукує нефротоксичний моноклональний імуноглобулін, що призводить до специфічного ураження нирок із неухильним прогресуванням ренальної дисфункції та погіршенням прогнозу хвороби [2].
Під моноклональною гаммапатією (МГ) розуміють наявність аберантного клону В-клітинної лінії диференціювання, що продукує молекулу імуноглобуліну (ІГ) або її частини [3].
Клон — популяція клітин, що виникла з однієї клітини-попередниці, яка успадковує всі її властивості, у тому числі здатність продукувати моноклональний парапротеїн. Моноклональний білок (парапротеїн, або М-протеїн) може мати патологічні властивості, що реалізуються різними шляхами, включаючи депозицію в органах і тканинах, призводячи до їх пошкодження. Клональні клітини мають можливість продукувати повнорозмірну молекулу ІГ або його частину (тільки легкий ланцюг (ЛЛ) або тільки важкий ланцюг). Залежно від етапу диференціювання В-клітин клональну проліферацію можна розділити: 1) на лімфоцитарну; 2) лімфоплазмоцитарну; 3) плазмоклітинну [4–7].
Клінічні прояви МГ пов’язані: а) зі збільшенням пухлинної маси; б) патологічними ефектами ІГ.
Моноклональні гаммапатії характеризуються секрецією цілого моноклонального імуноглобуліну або його фрагменту з популяції В-клітин, лімфоцитарного або плазмоцитарного походження. Іноді виділений моноклональний ІГ відповідає за пошкодження органів, проте найчастіше уражається нирка.
Широкий спектр нефропатій асоціюється з моноклональними гаммапатіями, проте переважно уражаються клубочки. За останні три десятиліття спектр моноклональних ІГ-зв’язаних нефропатій розширився із визначенням нових форм, включаючи хворобу відкладення важких ланцюгів (ХВВЛ), гломерулопатію С3, тромботичну мікроангіопатію та проліферативний гломерулонефрит (ГН) із відкладеннями депозитів моноклональних імуноглобулінів (МПГНМІД) [7–9]. Сучасна класифікація моноклональних гаммапатій ренального значення подана в табл. 1.
/51.jpg)
Ниркові ускладнення, пов’язані з моноклональними ІГ, зазвичай виникають у пацієнтів із млявим перебігом гематологічних порушень, що не відповідають діагностичним критеріям симптоматичної множинної мієломи або B-клітинної лімфоми та зазвичай проявляються аналогічно моноклональній гаммапатії невизначеного значення (МГНЗ). Це вказує на те, що нефротоксичність виникає не через швидкість секреції ІГ, що відображає пухлинне навантаження, а, скоріше, через внутрішні патогенні властивості самого моноклонального ІГ. Винятком є нефропатія з відкладенням легких ланцюгів, що незмінно виникає в контексті множинної мієломи великої маси (рідше — макроглобулінемії Вальденстрема) та зазвичай супроводжується характерними для мієломи синдромами CRAB (С — гіперкальціємія (hypercalcemia), R — ураження нирок (renal insufficiency), A — анемія (anemia), B — ураження нирок (bone lesions) [11, 18].
Дані клінічних досліджень щодо різних типів захворювань нирок, пов’язаних із моноклональними ІГ, демонструють, що пригнічення токсичних ІГ зазвичай призводить до поліпшення ниркових симптомів, а іноді й до повного зникнення, якщо лікування розпочате до розвитку необоротних фіброзних змін [12].
До недавнього часу через відсутність критеріїв симптоматичного гематологічного захворювання пацієнти з моноклональними ІГ-асоційованими нефропатіями часто зазнавали труднощів з отриманням відповідної хіміотерапії. Помітний прогрес у лікуванні було досягнуто з уведенням концепції моноклональної гаммапатії ренального значення, яка диференціює клінічні ситуації розвитку нефропатій, викликаних небезпечними невеликими клонами B-клітин, від ситуації МГНЗ, що не характеризується будь-яким пошкодженням органів-мішеней [2].
Нещодавно було введено термін «моноклональна гаммапатія клінічного значення» (МГКЗ). МГКЗ охоплює всі типи уражень органів, викликаних моноклональними ІГ, що продукуються безсимптомними клонами малих В-клітин, які не мають критеріїв для хіміотерапії, через прямі (відкладення в тканини) або непрямі механізми, включаючи активність автоантитіл, утворення імунного комплексу або активацію комплементу [11].
Патоморфологічна класифікація МГРЗ-нефропатій розрізняє ураження нирок (клубочкові або тубулоінтерстиціальні) за складом відкладень та їх ультраструктурним видом. Гломерулярні пошкодження:
— гломерулопатії без моноклональних відкладень ІГ (тромботична мікроангіопатія та гломерулопатія С3), при яких моноклональні імуноглобуліни не викликають пошкодження шляхом свого відкладення, а, скоріше, активізують альтернативний шлях комплементу;
— гломерулопатії з організованими моноклональними відкладеннями ІГ, включаючи ІГ амілоїдоз, імунотактоїдний ГН та кріоглобулінемічний ГН;
— гломерулярні захворювання з неорганізованими моноклональними ІГ, переважно представлені моноклональним відкладанням ІГ типу Рандалла (MIDD), що розділяється на хворобу відкладення легких ланцюгів (ХВЛЛ), хворобу відкладення важких ланцюгів та хворобу легких та важких ланцюгів. Проліферативний гломерулонефрит (ГН) з відкладеннями депозитів моноклональних імуноглобулінів (ПГНМІД) є останнім описаним типом у цій групі.
Проліферативний гломерулонефрит із відкладеннями депозитів моноклональних імуноглобулінів — нещодавно описане захворювання у спектрі моноклональної гаммапатії ренального значення. Патологічний процес обмежується нирками і проявляється хронічною хворобою клубочків, порушенням функції нирок та альбумінурією, іноді нефротичного рівня [13].
Морфологічно ПГНМІД характеризується переважно мембрано-проліферативним ГН, рідше зустрічаються ендокапілярний ГН, мезангіопроліферативний ГН та атиповий мембранозний ГН [8, 9].
ПГНМІД є рідкісним захворюванням, яке переважно описується у представників європеоїдної раси, без гендерних відмінностей. Частота морфологічних верифікацій коливається від 0,17 до 3,7 % [8, 14], що у вісім разів нижче, ніж при амілоїдозі з відкладенням імуноглобулінів легких ланцюгів (AL), і у два рази нижче, ніж при ІГ типу Рандалла (Randall-type monoclonal Ig deposition disease, MIDD) [15]. Середній вік на момент захворювання становить 55 років, але хвороба може виникнути в будь-якому віці. Близько 20 % пацієнтів старші від 70 років. У пацієнтів віком понад 20 років частота ПГНМІД при біопсії оцінюється в 0,8 %, переважно спостерігаються монотипні відкладення IgG3 [16].
ПГНМІД у більшості випадків є виключно нирковим захворюванням, без екстраниркових уражень, із депозицією монотипним ІГ в клубочках. Ниркові прояви неспецифічні та різного ступеня вираженості [8, 17]. На момент біопсії протеїнурія зазвичай постійна, складається переважно з альбуміну та становить > 3 г/добу у 70 % пацієнтів. Майже половина пацієнтів мають нефротичний синдром, а у 60 % визначається дифузний набряк. Гематурія, іноді макроскопічна, виявляється у 80 % випадків. Дві третини пацієнтів мають ниркову недостатність, із вихідною середньою ШКФ 36 мл/хв/1,73 м2, < 10 % хворих потребують діалізу на момент встановлення діагнозу [8]. Швидко прогресуючий ГН є рідкісним та вказує на тяжкі ендокапілярні та екстракапілярні проліферативні ураження.
Клінічний випадок мезангіопроліферативного гломерулонефриту з депозитами моноклональних імуноглобулінів
Пацієнтка О., 35 років, звернулась на консультацію зі скаргами на периферичні набряки, виражену слабкість, задишку, зниження темпу діурезу, підвищення артеріального тиску. Вважає себе хворою з серпня 2019 року, коли після переохолодження з’явились ознаки циститу. Лікувалася амбулаторно в лікаря-уролога, із позитивною динамікою. З вересня з’явилися набряки гомілок, які швидко поширювалися по всіх кінцівках. Була госпіталізована до Інституту терапії імені Л.Т. Малої 17.10.2019, де після обстеження встановлений діагноз «гострий гломерулонефрит. Нефротичний синдром». Призначено лікування: мікофенолату мофетил 2 грами з 21.10.2019. На тлі терапії білок у сечі знизився з 9 до 1 г/л. Пацієнтка консультована гематологом (проведена стернальна пункція, дані за мієлому відсутні), білок Бенс-Джонса в сечі +++. При динамічному спостереженні стан пацієнтки був стабільним, протеїнурія 1–1,5 г/добу. Додатково було призначено метилпреднізолон 8 мг, із позитивним ефектом, до повної ліквідації протеїнурії. Через 3 місяці після відміни метилпреднізолону стався рецидив захворювання, стан погіршувався, наростав набряковий синдром, погіршення контролю артеріального тиску, зниження темпу діурезу, у зв’язку з чим звернулась на консультацію.
З анамнезу відомо: у пацієнтки у 12 років був вивих тазостегнового суглоба, після чого діагностований коксартроз, який ускладнився асептичним некрозом, що загоювався упродовж 2 років; опікова хвороба (36 % тіла) у 9-річному віці.
Об’єктивно: стан пацієнтки середньої тяжкості. Шкіра бліда, післяопікові шрами. Набряковий синдром — верхні та нижні кінцівки, тулуб, живіт, гідроторакс. Оцінка показників життєдіяльності: артеріальний тиск 150/90 мм рт.ст., ЧСС 100/хвилину, ЧДР 22/хвилину. Пальпаторно — у правій здухвинній ділянці пальпується нирка, помірно болісна.
Дані обстеження до лікування наведені в табл. 2.
Пацієнтці проведена нефробіопсія.
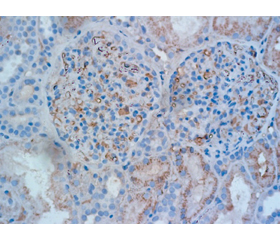
При патоморфологічному дослідженні виявлено: у біоптатах нирки до 12 клубочків, вони різного розміру, окремі збільшені за рахунок як дифузної проліферації мезангіальних клітин, набряку та сегментарної проліферації ендотеліальних клітин, так і фібриноїдних змін, в інших клубочках — нерівномірна (переважно від слабко до помірно вираженої) проліферація мезангіальних клітин, сегментарно — набряк ендотеліальних клітин, частина капілярних петель інфільтрована моноцитами, нерівномірно виражене збільшення мезангіального матриксу, в окремих клубочках невеликі ділянки сегментарного склерозу, які мають тенденцію до нодулярного характеру (рис. 2).
У частині капілярних петель набряк, у всіх клубочках нерівномірне, різного ступеня вираженості потовщення, у частині клубочків розщеплення базальних мембран капілярів, окремі капілярні петлі практично не змінені, точкові зрощення периферичних капілярних петель із капсулою Боумена, у 7 клубочках — уздовж всього периметра; в просторі капсули Боумена частини кубочків білкові маси, фібрин, нерівномірно виражене потовщення базальної мембрани капсули Боумена, в окремих клубочках виражене за рахунок склерозу (рис. 3).
У канальцях різко виражені дегенеративні зміни (за типом зернистої, вакуольної, на окремих ділянках гіаліново-краплинної дистрофії), некроз груп клітин епітелію, ділянки атрофії епітелію з потовщенням тубулярної базальної мембрани (до 10 % об’єму біоптату). У просторі канальців — білкові маси, злущений епітелій, просвіт частини канальців різко зменшений та/або відсутній за рахунок виражених дегенеративних змін в епітелії (рис. 1, 2).
У стромі — набряк, ділянки інтерстиціального фіброзу, у тому числі й периваскулярного (до 10 % об’єму біоптату), вогнищеві лімфогістіоцитарні клітинні інфільтрати. У судинах різного калібру — потовщення стінки за рахунок дисмукоїдозу, в окремих — ознаки фібриноїдного некрозу. При забарвленні конго червоним негативна реакція (рис. 4).
Імуногістохімічне дослідження: депозити IgA (переважання) (рис. 5), IgG, IgM, С3: у клубочках нерівномірні сегментарні гранулярні депозити в мезангіальній зоні, уздовж ендотелію капілярів від + до ++, у стромі між канальцями; у перитубулярних капілярах депозитів від + до ++, в епітелії частини канальців депозити у вигляді зернистості від + до ++. С1q: негативна експресія. CD20 (В-лімфоцити): відсутні позитивні В-лімфоцити. АА-амілоїд: негативна експресія в клубочках, стінках судин, стромі.
Легкі ланцюги імуноглобуліну типу κ та λ: депозити κ-легких ланцюгів імуноглобуліну(переважання): у клубочках сегментарні гранулярні депозити вздовж капілярних петель від + до ++, в окремих потовщених капілярних петлях до +++, депозити в мезангіальній зоні від + до ++, вогнищево депозити в стромі в перитубулярних капілярах від + до ++, в епітелії канальців депозити у вигляді зернистості від ++ до +++ (рис. 6, 7).
Патоморфологічний діагноз: мезангіопроліферативний гломерулонефрит (із переважанням депозитів IgА, меншою мірою IgG, IgM, C3-фракції комплементу) з вираженим тубулярним та слабко вираженим інтерстиціальним компонентом, який потребує диференціальної діагностики щодо змін у нирках при неамілоїдній формі відкладення моноклональних імуноглобулінів — хворобі легких ланцюгів (Ligh Chain Deposition Disease), яка входить до числа хвороб із моноклональною депозицією імуноглобулінів (MIDD, Monoclonal Immunoglobulin Deposition Disease).
Ступінь хронізації (Sethi et al., 2017) — СG 2 (mild chronic changes, 2 бали): гломерулосклероз (GS) 0, інтерстиціальний фіброз (ІФ) 1, тубулярна атрофія (TA) 1, артеріолосклероз (CV) 0.
Після отримання даних нефробіопсії додатково призначено визначення парапротеїну в крові і сечі (табл. 3).
Враховуючи дані обстежень, був установлений діагноз: ХХН І ст.: мезангіопроліферативний гломерулонефрит (IgA, IgM, IgG) із депозитами моноклональних імуноглобулінів (ЛЛ типу лямбда та каппа). Нефротичний синдром. Хвороба легких ланцюгів.
Додатково рекомендована консультація гематолога, консультація кардіолога (із метою виключення ураження серця).
Призначена терапія: метилпреднізолон 48 мг/добу; пульс-терапія циклофосфамідом 1 г/місяць, № 3, ацетилсаліцилова кислота 75 мг/дoбу, валсартан 40 мг, торасемід 10 мг/добу.
На тлі терапії стан пацієнтки з позитивною динамікою. Повний регрес набрякового синдрому, артеріальний тиск стабільний, не перевищує 120/80 мм рт.ст. Динаміка лабораторних показників під час терапії подана в табл. 4. Пацієнтка консультована кардіологом, ураження серця виключене. Консультація гематолога: хвороба легких ланцюгів. Рекомендовано: трепанобіопсія кісткового мозку, ректальна біопсія, проте враховуючи, що пацієнтка отримує терапію, результати можуть бути не інформативними. Контроль парапротеїну крові та сечі в динаміці терапії. Як терапія (після отримання результатів дослідження) можуть бути розглянуті: бортезоміб, імуномодуляторні ліки, ВДХТ + АТГСК.
Висновки
1. Моноклональна гаммапатія ренального значення — це не самостійне захворювання нирок, не хронічний гломерулонефрит, а стан, при якому ураження нирок вторинне щодо клональної В-клітинної проліферації. Іншими словами, МГРЗ — передпухлинне захворювання в поєднанні з хронічною хворобою нирок, що потребує негайного початку лікування.
2. Проліферативний гломерулонефрит із відкладеннями депозитів моноклональних імуноглобулінів — нещодавно описане захворювання у спектрі моноклональної гаммапатії ренального значення. Патологічний процес обмежується нирками і проявляється хронічною хворобою клубочків, порушенням функції нирок та альбумінурією, іноді нефротичного рівня. Діагностика моноклональних ІГ-асоційованих нефропатій часто зазнає труднощів, у зв’язку з чим нефробіопсія вирішальна у встановленні діагнозу.
3. Саме мультидисциплінарний підхід, командна робота гематологів, морфологів та нефрологів забезпечить найкращі прогнози, у тому числі і ниркові, та дозволить запобігти прогресуванню хронічної хвороби нирок до термінальних стадій.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці статті.
Отримано/Received 05.01.2022
Рецензовано/Revised 13.01.2022
Прийнято до друку/Аccepted 20.01.2022
Список литературы
1. Leung N., Bridoux F., Batuman V. et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019. 15(1). 45-59. doi: 10.1038/s41581-018-0077-4.
2. Leung N., Bridoux F., Hutchison C.А. et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012. 120. 4292-4295.
3. Bridoux F., Leung N., Hutchison C.А. et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015. 87. 698-711.
4. Dimopoulos M.А., Sonneveld P., Leung N. et al. International Myeloma Working Group Recommendations for the Diagnosis and Management of Myeloma-Related Renal Impairment. J. Clin. Oncol. 2016. 34(13). 1544-1557. doi: 10.1200/JCO.2015.65.004.
5. Hutchison C.А., Batuman V., Behrens J. et al. The pathogenesis and diagnosis of acute kidney injury in multiple myeloma. Nat. Rev. Nephrol. 2011. 8. 43-51.
6. Strati P., Nasr S.Н., Leung N. et al. Renal complications in chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis: the Mayo Clinic experience. Haematologica. 2015. 100. 1180-1188.
7. Touchard G. Ultrastructural pattern and classification of renal monoclonal immunoglobulin deposits. In: Touchard G., Aucouturier P., Hermine O., Ronco P. (eds). Monoclonal Gammopathies and the Kidney. Dordrecht, Netherlands: Kluwer Academic Publishers, 2003. 95-120.
8. Nasr S.Н., Satoskar A., Markowitz G.S. et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J. Am. Soc. Nephrol. 2009. 20. 2055-2064.
9. Bridoux F., Javaugue V., Nasr S.H., Leung N. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: a nephrologist perspective. Nephrol. Dial. Transplant. 2021. 36. 208-215 doi: 10.1093/ndt/gfz176.
10. Merlini G., Stone M.J. Dangerous small B-cell clones. Blood. 2006. 108 (8). 2520-2530. doi: 10.1182/blood-2006-03-001164.
11. Fermand J.Р., Bridoux F., Dispenzieri A. et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018. 132(14). 1478-1485. doi: 10.1182/blood-2018-04-839480.
12. Ohashi R., Sakai Y., Otsuka T. et al. Proliferative glomerulonephritis with monoclonal IgG2j deposit successfully treated with steroids: a case report and review of the literature. CEN Case Rep. 2013. 2. 197-203.
13. Alpers C.Е., Tu W.Н., Hopper J. et al. Single light chain subclass (kappa chain) immunoglobulin deposition in glomerulonephritis. Hum. Pathol. 1985. 16. 294-304.
14. Gowda K.K., Nada R., Ramachandran R. et al. Proliferative glomerulonephritis with monoclonal immunoglobulin deposition disease: the utility of routine staining with immunoglobulin light chains. Indian J. Nephrol. 2015. 25. 344-348.
15. Nasr S.Н., Markowitz G.S., Stokes M.В. et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immunecomplex glomerulonephritis. Kidney Int. 2004. 65. 85-96.
16. Xing G., Gillespie R., Bedri B. et al. Proliferative glomerulonephritis with monoclonal IgG deposits in children and young adults. Pediatr. Nephrol. 2018. 33. 1531-1538.
17. Bhutani G., Nasr S.Н., Said S.М. et al. Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clin. Proc. 2015. 90. 587-596.
18. Merlini G., Stone M.J. Dangerous small B-cell clones. Blood. 2006. 108 (8). 2520-2530. doi: 10.1182/blood-2006-03-001164.

/51.jpg)
/52.jpg)
/50.jpg)
/53.jpg)
/54_2.jpg)
/54.jpg)
/55.jpg)