Резюме
Актуальність. Бічний аміотрофічний склероз (БАС) — це захворювання ділянок нервової системи, які конт-ролюють довільні рухи м’язів. При БАС рухові нейрони (нервові клітини, що контролюють м’язові клітини) поступово втрачаються. При цьому м’язи, якими вони керують, стають слабкими, а потім нефункціональними, що призводить до м’язової слабкості, інвалідності та зрештою смерті. БАС є найпоширенішою формою хвороби моторних нейронів. У недавніх дослідженнях продемонстровано, що схема прогресування в пацієнтів зі спорадичним БАС, а також те, які фактори впливають на прогресування патології, залишається нерозгаданою таємницею. Тому ми провели дослідження з використанням переглянутої функціональної шкали БАС (ALSFRS-R) та вивчили фактори, що сприяли його швидкому прогресуванню, у Першій університетській клініці Тбіліського державного медичного університету в 2019–2022 роках. Матеріали та методи. Обстежено 60 хворих на БАС: 31 чоловік (51,67 %) та 29 жінок (48,33 %) віком 21–84 роки. Магнітно-резонансну томографію головного мозку виконано в пацієнтів, які приймали рилузол, осіб зі спорадичним БАС, електрофізіологічні дослідження проводили усім хворим. Діагноз встановлювали за критеріями Gold Coast, пацієнтів обстежували з використанням шкали ALSFRS-R. Опитування щодо факторів, які впливають на прогресування захворювання, проводилося за допомогою опікуна пацієнта (розроблено спеціальну анкету для оцінки ймовірних модифікаторів прогресування захворювання). Результати. Виявлено, що нещодавні падіння, травми, хірургічні втручання та інфекції мають значний вплив на прогресування захворювання. Було встановлено, що БАС є нелінійно прогресуючим захворюванням і може варіюватися. Ми виявили, що в 5 % пацієнтів спостерігалося покращення за шкалою ALSFRS-R, що не пояснювалося ні прийомом ліків, ні зміною способу життя. Висновки. Швидкість прогресування БАС сама по собі залишається загадкою. Прогнозування швидкості прогресування та факторів, що впливають на нього, було б корисним для пацієнтів з БАС і навіть може дати шанс уповільнити або зупинити прогресування. Необхідні подальші дослідження.
Background. Amyotrophic lateral sclerosis (ALS) is a disease of the parts of the nervous system that control voluntary muscle movement. In ALS, motor neurons (nerve cells that control muscle cells) are gradually lost. As this happens, the muscles they control become weak and then nonfunctional, thus leading to muscle weakness, disability, and eventually death. ALS is the most common form of motor neuron disease. Recent studies widely demonstrated that the pattern of progression in sporadic ALS patients is unsolved mystery, especially what factors influence the progression of the disease. Therefore, we conducted a study using ALS Functional Rating Scale-Revised (ALSFRS-R) for disease progression and investigated factors contributing to its rapid rate in The First University Clinic of Tbilisi State Medical University in 2019–2022. Materials and methods. A total of 60 patients with ALS were examined, among them 31 male (51.67 %), 29 female (48.33 %) aged 21–84 years. Riluzole takers, patients with sporadic ALS underwent brain magnetic resonance imaging, and electrophysiological studies were done in all patients. Participants were diagnosed using the Gold Coast criteria and examined with the ALSFRS-R. Survey for factors affecting disease progression was performed with a help of the patient’s caregiver (a disease-specific questionnaire was invented for this purpose, investigating probable progression modifiers). Results. It has been found that recent falls, trauma, surgery, and infection have a drastic impact on disease progression. ALS was found to be a not linear progressive disease and can vary. We have found that 5 % of patients had improvement оn ALSFRS-R, not explained by medication, nor lifestyle change. Conclusions. The rate of ALS progression appears to be a mystery by itself. Predicting the progression rate, and the factors affecting it would be beneficial for ALS patients and may even bring chances to slow or halt the progression. Further research is essential.
Introduction
Amyotrophic lateral sclerosis (ALS) is a disease of the parts of the nervous system that control voluntary muscle movement. In ALS, motor neurons (nerve cells that control muscle cells) are gradually lost. As this happens, the muscles they control become weak and then nonfunctional, thus lea-ding to muscle weakness, disability, and eventually death. ALS is the most common form of motor neuron disease [5–9].
Amyotrophic lateral sclerosis is one of the severe neurodegenerative diseases [6–9], a progressive neuromuscular disease, first records of which have been made in medical literature in the first half of 19th century (Charles Bell, 1824; François-Amilcar Aran, 1850) [1–4].
The clinical symptoms of the disease, in line with developed neurological deficit, were described by Jean-Martin Charcot in 1869. He also was the one who coined the term “amyotrophic lateral sclerosis”. This disease was very rare in 19th century and there were only a few methods developed for its diagnosis [7–10].
Recent studies widely demonstrate that the pattern of progression in sporadic ALS patients is unsolved mystery, especially what factors influence the progression of the disease. Therefore, we conducted a study using ALS Functional Rating Scale-Revised (ALSFRS-R) for disease progression and investigated factors contributing to its rapid rate in The First University Clinic of Tbilisi State Medical University in 2019–2022.
Materials and methods
A total of 60 patients with ALS were examined, 31 male (51 %), 29 female (49 %) aged 21–84 years.
Pedigree analysis was also performed: a pedigree was compiled using standard symbols, which includes information on the health status of the proband’s relatives. As a result, relatives who are at risk of developing the disease or are carriers of the pathogenic allele have been identified. Such individuals were given appropriate genetic counseling and recommended for genetic testing.
Results
Upon necessity, the risk of developing the disease for the next generation was calculated. Five (8.33 %) patients had confirmed diabetes mellitus, one (1.67 %) had a family history of athetosis, 4 (6.67 %) had to contact with poisonous chemicals, 2 (3.33 %) have a history of brain trauma, 35 patients (58.33 %) were tobacco users.
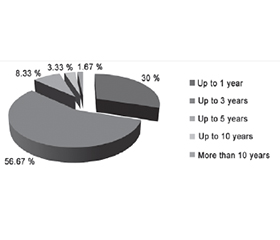
The duration of ALS was as follows: up to 1 year — in 18 (30 %) patients, up to 3 years — in 34 (56.67 %), up to 5 years — in 5 (8.33 %), up to 10 years — 2 (3.33 %), more than 10 years — in 1 (1.67 %) (Fig. 1).
Patients’ functional condition was evaluated by the –ALSFRS according to the following symptoms: speech, salivation, swallowing, writing, chewing, and processing food (with or without a gastrostomy), dressing and hygiene, tur-ning in bed and dressing, walking, stair-climbing, breathing.
The higher the total score, the more the patient’s vital functions are preserved.
Mild functional changes (31 to 40 points according to ALSFRS) were found in 14 (23.33 %) patients, moderate severity at the time of examination (21 to 30 points) was detected in 15 (25 %) cases, severe form (10 to 20 points) — in 25 (41.67 %) patients, and 6 (10 %) participants had very severe form (0 to 10 points), which is shown in Fig. 2.
The disease started with symptoms such as numbness of muscles, weakness of limbs, fasciculations in 36 (60 %) patients.
According to our research, the diagnosis of ALS is difficult until muscle atrophy and tremors are detected. In 23 (38.33 %) cases, ALS started with symptoms on one or both legs. Patients felt uncomfortable while walking, the ankle lost its flexibility, its range of motion was limited. Muscle weakness was expressed, muscle spasms, increase of deep reflexes or expansion of the reflexogenic zone, pathological reflexes, pronounced muscle atrophy, increased spasticity were also detected. At this time, the upper limbs were less damaged, although the flexibility of the fingers was limited.
Bulbar-onset ALS was detected in 24 (40 %) patients with difficulty speaking, the patient spoke “through the nose”, later had difficulty swallowing. Speech disorders (dysarthria, anarthria), voice production disorders (dysphonia, aphonia), disappearance of gag reflex, salivation, breathing disorders soon added to the symptoms. In 11 (18.33 %) patients with ALS confirmed by us, the symptoms included signs of both lower and upper motor neuron lesions. Upper motor neuron lesion manifested in muscle hypertonia, hyperreflexia, pathological Babinski sign. In case of damage to the lower motor neurons, muscle weakness and atrophy, fasciculations were present. According to our study, patients lost their ability to move independently, but their mental abilities were not limited. They survive through artificial lung ventilation and artificial nutrition.
Riluzole takers, patients with sporadic ALS underwent brain magnetic resonance imaging, and electrophysiological studies were done in all patients. Participants were diagnosed using the Gold Coast criteria and examined with the ALSFRS-R. Survey for factors affecting disease progression was performed with a help of the patient’s caregiver (a disease-specific questionnaire was invented for this purpose, investigating probable progression modifiers).
Conclusions
It has been found that recent falls, trauma, surgery, and infection have a drastic impact on disease progression. ALS was found to be a not linear progressive disease and can vary. We have found that 5 % of patients had improvement on –ALSFRS-R, not explained by medication, nor lifestyle changes.
The rate of ALS progression appears to be a mystery by itself. Predicting the progression rate, and the factors affec-ting it would be beneficial for ALS patients and may even bring chances to slow or halt the progression. Further research is essential.
Received 03.03.2024
Revised 14.03.2024
Accepted 23.03.2024
Список литературы
1. Kekenadze M, et al. WE-150. Role of neuromuscular junction discharges in the pathophysiology of ALS. Clinical Neurophysiology. 2022;141:S74.
2. Kekenadze M, Rocca C, Kaiyrzhanov R, Nagy S, Kvirkvelia N, et al. Analysis of C9orf72 repeat expansions in Georgian patients with ALS. F1000Res. 2024 Mar 6;12:1113. doi: 10.12688/f1000research.138436.1.
3. Kekenadze M, Beridze M, Kvirkvelia N, Vashadze S. Anxiety — prognostic marker of ALS? European Journal of Neurology. 2022;29(1):EPV-529.
4. Kekenadze M, et al. Clinical characteristics of ALS in Georgian patients. Georgian Medical News. 2021;319:71-75.
5. Monarch Disease Ontology, released 2018-06-29.
6. Stanford Medicine School of Medicine. Epidemiology of Sporadic ALS. 2015.
7. Al-Chalabi A, Leigh PN. Recent advances in amyotrophic late-ral sclerosis. Current Opinion in Neurology. 2000 Aug;13(4):397-405. doi: 10.1097/00019052-200008000-00006.
8. Garnier C, Devred F, Byrne D, Puppo R, Roman AYu. Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Scientific Reports. 2017;7:6812. doi: 10.1038/s41598-017-07215-7.
9. Caragounis A, Price KA, Soon CPW, Filiz G, Masters CL. Zinc induces depletion and aggregation of endogenous TDP-43. Free Radical Biology and Medicine. 2010 May 1;48(9):1152-61. doi: 10.1016/j.freeradbiomed.2010.01.035.
10. ALSUntangled Group. ALSUntangled Update 1: Investigating a bug (Lyme Disease) and a drug (Iplex) on behalf of people with ALS. Amyotrophic Lateral Scler. 2009 Aug;10(4):248-50. doi: 10.1080/17482960903208599.

/37.jpg)