Статтю опубліковано на с. 63-69
Гіпопітуїтаризм — захворювання, що виникає внаслідок відсутності або неадекватності секреції одного або декількох гормонів передньої частки гіпофіза. Відомо, що в аденогіпофізі синтезуються й виділяються в системний кровообіг такі гормони, як соматотропний гормон (СТГ), адренокортикотропний гормон (АКТГ), лютеїнізуючий гормон (ЛГ), фолікулостимулюючий гормон (ФСГ), тиреотропний гормон (ТТГ) та пролактин, дефіцит яких призводить до зниження функції відповідних ендокринних залоз (кори надниркових залоз, яєчок або яєчників, щитоподібної залози). Тому клінічні прояви хвороби обумовлені відсутністю дії на організм гормонів означених залоз. Дефіцит СТГ проявляється пропорційною затримкою росту дітей та гіпоглікеміями, дефіцит гонадотропінів призводить до затримки статевого розвитку, а в разі вродженого гіпопітуїтаризму — до двобічного крипторхізму та пренатальної мікропенії; АКТГ — до втрати маси тіла, гіпоглікемії (внаслідок відносного гіперінсулінізму), втомлюваності, гіпотензії та іноді навіть до смерті; недостатня секреція ТТГ обумовлює клініку гіпотиреозу. Проте домінуючим в клініці гіпопітуїтаризму у дітей є симптом затримки росту (гіпофізарний нанізм) та/або його темпів. Поширеність дефіциту СТГ серед дітей у світі становить від 1 : 4000 до 1 : 20000 [1, 2]. Існує декілька класифікацій гіпопітуїтаризму, одна з них [3] поділяє гіпопітуїтаризм на вроджений або набутий.
Класифікація гіпопітуїтаризму
І. Вроджений:
1. Спадковий:
— Ізольований дефіцит СТГ:
А. Мутації гена СТГ (GH‑1):
1) тип IA: рівень СТГ не визначається (делеція гена СТГ, автосомно-рецесивний тип спадкування (Illig-тип СТГ-дефіциту)
2) тип IБ: рівень СТГ знижений (автосомно-рецесивний тип спадкування)
3) тип II: рівень СТГ знижений (автосомно-домінантний тип спадкування)
4) тип III: Х-зчеплена рецесивна форма спадкування. При цьому типі мутації в GH‑1 відсутні; окрім низькорослості пацієнти можуть мати агаммаглобулінемію.
Б. Мутації гена рецептора до СТГ-рилізинг гормона (GHRH-R).
— Множинний дефіцит гормонів аденогіпофіза:
1) мутації гена PІT1;
2) мутації гена PROP1 та ін.
2. Ідіопатичний вроджений дефіцит СТГ-РГ.
3. Дефекти розвитку гіпоталамо-гіпофізарної системи (патологія серединної трубки (ан-, голопрозенцефалія, септооптична дисплазія) і дизгенезія гіпофіза (вроджена аплазія, гіпоплазія, ектопія гіпофіза) тощо.
II. Набутий:
— пухлини гіпоталамуса й гіпофіза, інших відділів мозку, супраселярні арахноїдальні кісти;
— травми, інфекції головного мозку, гідроцефалія;
— симптом порожнього турецького сідла;
— патологія судин головного мозку;
— опромінення голови й шиї в анамнезі, токсичні наслідки хіміотерапії, інфільтративні хвороби тощо.
В основі іншої класифікації лежить ступінь ураження гіпофіза:
— пангіпопітуїтаризм — недостатність секреції всіх гормонів гіпофіза;
— парціальний гіпопітуїтаризм — недостатність секреції декількох гормонів гіпофіза;
— селективний (ізольований) гіпопітуїтаризм — недостатність секреції одного гормона гіпофіза.
Найбільш важливим питанням для практикуючого лікаря є своєчасне діагностування гіпопітуїтаризму в дітей, у якому можна виділити такі напрямки:
1. Клініко-анамнестичний. Ретельно зібраний анамнез враховує ріст та масу тіла дитини при народженні, швидкість росту (що має становити не менше від 6 cм/рік у дітей віком 2–4 років, не менше від 5 cм/рік — у дітей віком 4–8 років, а в дітей препубертатного віку — близько 4 см/рік), а також ступінь постнатального відставання росту (субнанізм/нанізм, які діагностують при затримці росту понад 2 стандартні відхилення (> (–) 2,0 SDS) або нижче 10-го перцентиля відповідно до діаграм росту ВООЗ (WHO Growth Charts) або Центру з контролю та профілактики захворювань (CDC Growth Charts) [4].
2. Гормональний, що включає визначення базального вмісту гормонів: СТГ, інсуліноподібного фактора росту 1 (ІФР‑1), інсуліноподібного фактора росту 3 (ІФР‑3) та протеїну, що зв’язує ІФР‑3 (ІФРЗБ‑3), ФСГ, ЛГ, АКТГ, кортизолу в крові, слині й добовій сечі, ТТГ, вільного тироксину (вТ4), а також проведення стимуляційних проб.
3. Генетичний, у тому числі визначення каріотипу.
4. Рентгенологічний (визначення кісткового віку з оцінкою за атласом Greulich and Pyle [5]).
5. Інструментальні методи (магнітно-резонансна томографія (МРТ) головного мозку, гіпофіза; ультразвукове дослідження (УЗД) гонад).
Домінуючою в клініці гіпопітуїтаризму в дітей є затримка росту різного ступеня (гіпофізарний нанізм). Слід зауважити, що ізольована оцінка фізіологічної секреції СТГ дитини (базальний рівень, спонтанна нічна секреція, дослідження секреції під час фізичного навантаження тощо) не можуть слугувати діагностичними критеріями СТГ-недостатності, тому для її підтвердження необхідно проводити стимуляційні тести (з клофеліном, інсуліном, аргініном, глюкагоном тощо). Оскільки жоден тест не має 100% доказовості, для більш вірогідного діагнозу слід проводити не менше від двох стимуляційних тестів з визначенням рівня СТГ у різних часових точках (табл. 1).
/64.jpg)
Стимуляційні тести засновані на здатності різних фармакологічних препаратів стимулювати секрецію й викид СТГ соматотрофами аденогіпофіза. Обов’язковою умовою проведення стимуляційних тестів є стан еутиреозу (компенсація первинного або вторинного гіпотиреозу). У різних країнах застосовують різні стимуляційні тести, зокрема в Україні найчастіше застосовують тести з клофеліном та інсуліном (табл. 1), а, наприклад, у Великобританії — тести з глюкагоном та аргініном [1].
Під час проведення стимуляційних тестів можна отримати некоректні результати за умови наявності в дитини некомпенсованого гіпотиреозу (первинного, вторинного), несприятливих психосоціальних чинників розвитку дитини (незадовільне харчування, погані стосунки в сім’ї), у хворих з ожирінням, затримкою статевого розвитку в препубертатному періоді, під час вживання глюкокортикоїдів або психотропних засобів (табл. 2).
Також одним із важливих чинників, що впливають на результати функціональних проб, є використання різних лабораторних методів для визначення СТГ. Так, застосування лабораторіями різних методів дає коливання показників СТГ від 21 до 57 % залежно від використаного методу [7]. Тому вимірювання СТГ методом мас-спектрометрії має значні переваги порівняно з методом імуноаналізу, оскільки він є повністю незалежним від концентрації в сироватці крові СТГ-зв’язуючого протеїну [8].
Також найбільш часто хибнонегативні результати можна отримати у підлітків з конституційною затримкою росту, у яких спостерігається низька секреція стимуляційного СТГ в препубертаті або на початку статевого дозрівання, але в пубертатному періоді викиди СТГ знаходяться в межах норми. Зниження викиду СТГ у таких дітей може бути обумовлене відсутністю статевих гормонів на момент первинного тестування. Для уникнення гіпердіагностики гіпосоматотропізму (і, відповідно, значних фінансових витрат державою та потенційних побічних реакцій на лікування) в багатьох країнах перед проведенням стимуляційних тестів застосовують steroid priming (праймінг, тобто попередній курс статевих стероїдів) [6, 9]. Зокрема, він може бути може бути рекомендований для підлітків із затримкою статевого розвитку (у дівчаток віком > 11–12 років і хлопчиків віком > 13–13,5 року) [9]. Інші автори застосовують праймінг у всіх дітей препубертатного періоду (для дівчаток віком > 8 років і хлопчиків віком > 9 років) [1]. При цьому хлопчикам застосовують препарати тестостерону депо 100 мг внутрішньом’язово за 5 днів до стимуляційних тестів, а дівчаткам — етинілестрадіол 40 мкг/м2 або 25 мг двічі на добу 5 днів (або β-естрадіол 2 мг/добу 3 дні). Використання праймінгу дозволяє знизити гіпердіагностику дефіциту СТГ у здорових препубертатних дітей з 39 до 5 % [10].
Рівень ІФР‑1 також може бути знижений не тільки при дефіциті СТГ, а й при психосоціальній депривації, недоїданні, гіпотиреозі, патології печінки та нирок. Для дітей віком до трьох років більш інформативним є визначення ІФР‑3/ІФРЗБ‑3. Оцінювати результат ІФР слід шляхом застосовування перцентильних таблиць (за Cole and Green, 1992; Yee, 2004) або за стандартним відхиленням (SD), що розраховується відповідно до віку і статі. Для встановлення діагнозу гіпофізарного нанізму рівень ІФР‑1 має бути знижений більше двох стандартних відхилень (> (–) 2,0 SD), що переконливо вказує на дефіцит гормона росту у випадку, якщо інші причини низького ІФР‑1 були виключені (розрахунок ІФР див. на http://www.esoterix.com/endocrinology-services/endocrinology-tools/calculator-igf1). Рівень ІФР‑1 може бути також знижений у 25–50 % дітей з ідіопатичною низькорослістю, однак нормальний або незначно знижений рівень ІФР‑1 ставить під сумнів, хоча й і не виключає дефіцит СТГ.
У нормі при проведенні стимуляційних тестів викиди СТГ мають перевищувати 10 нг/мл. Якщо максимальний викид СТГ на стимуляцію становить < 10,0 нг/мл, у дитини є дефіцит СТГ; якщо викид < 7,0 нг/мл — діагностують тотальний дефіцит СТГ; при викиді від 7,0 до 10,0 нг/мл йдеться про парціальний дефіцит СТГ. Якщо при проведенні першого стимуляційного тесту рівень СТГ > 10 нг/мл, другий стимуляційний тест не проводиться. З іншого боку, за наявності у дитини множинного дефіциту гормонів гіпофіза (при лабораторно підтвердженому вторинному гіпотиреозі, гіпокортицизмі тощо) достатньо проведення одного стимуляційного тесту [1, 11]. На практиці, як правило, першим застосовують клонідиновий тест, а потім — інсуліновий. Хоча інсуліновий тест вважають ще й золотим стандартом діагностики вторинного гіпокортицизму (поряд із стимуляційним тестом з аналогом АКТГ), його застосування пов’язують з розвитком ускладнень під час проведення (а саме розвитком тяжких гіпоглікемій, особливо небезпечних у дітей), тому він має низку протипоказань (табл. 2) і потребує проведення в умовах відділень спеціалізованих стаціонарів. Для виключення вторинного гіпокортицизму при проведенні інсулінового тесту концентрація кортизолу в будь-якій точці повинна вдвічі перевищувати базальну.
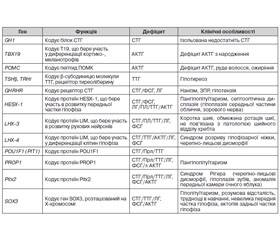
З розвитком молекулярної генетики виникла реальна можливість вивчення генетичних мутацій, що призводять до різних форм гіпопітуїтаризму [12], це дозволяє в деяких випадках точно встановити діагноз або визначити прогноз захворювання, провести медико-генетичну консультацію членів родини тощо (табл. 3).
/66.jpg)
Серед вищеописаних мутацій найчастішою причиною множинного дефіциту гормонів гіпофіза є мутація PROP1 [13–15]. Гормональний фенотип у пацієнтів з PROP1 мутаціями характеризується дефіцитом СТГ, ТТГ, пролактину, гонадотропних гормонів, АКТГ та найчастіше проявляється в дитячому й підлітковому віці [16–19]. Більшість дітей з такою мутацією мають нормальний зріст і масу тіла при народженні, у матерів був неускладнений перебіг вагітності [15]. Затримка росту відбувається вже у ранньому дитинстві, у той час як випадіння інших гормональних функцій зазвичай виявляють пізніше [16]. У таких пацієнтів не відбувається спонтанний пубертат, а випадіння АКТГ діагностують найпізніше. Найчастіше цю мутацію ідентифікують у вигляді двох варіантів — c.296delGA та c.150delA [13, 20–27]. Використання генетичного аналізу, зокрема підтвердження мутації PROP1, дозволяє проводити адекватний клінічний і гормональний моніторинг усіх тропних гормонів, в тому числі для уникнення запізнілої діагностики гіпокортицизму й ризиків, з цим пов’язаних [28]. Близько 20 % пацієнтів з мутаціями PROP1 мають гіперплазію аденогіпофіза за даними МРТ з подальшою його інволюцією в процесі життя, аж до розвитку картини порожнього турецького сідла. Раніше таку МРТ-картину гіперплазії аденогіпофіза розцінювали як пухлинний процес (краніофарингіома, аденома гіпофіза), що призводило часом навіть до необґрунтованих оперативних втручань на гіпофізі. Сьогодні подібна МРТ-картина в дитини будь-якого віку з дефіцитом СТГ/Прл/ТТГ є показанням для генетичної діагностики, насамперед аналізу гена PROP1.
Вторинний (гіпогонадотропний) гіпогонадизм
Вторинний гіпогонадизм — доволі непоширене захворювання, частота якого сягає 1 : 8000 у дівчаток і 1 : 4000 у хлопчиків [29]. Для підтвердження необхідна поглиблена гормональна діагностика (зокрема, проведення стимуляційних тестів). Клінічно наявність вторинного гіпогонадизму можна запідозрити при відсутності вторинних статевих ознак у дівчаток після 13 років, а у хлопчиків — після 14 років. Тут також виділяють вроджені та набуті форми [30].
Класифікація вторинного гіпогонадизму
1. Врождені форми:
— ізольовані форми: синдром Кальмана з аносмією або без неї;
— у складі дефіциту інших гіпофізарних гормонів: дефект PROP1, синдром фертильних євнухів, синдром Паскуаліні;
— при синдромальних патологіях: синдром Прадера — Віллі, Барде — Бідля, Лоренса — Муна, Рода, Медока;
— мозочкові атаксії з гіпогонадизмом (атаксія Фрідрейха, синдром Маринеско — Шегрена, синдром Луї-Бар, синдром Буше — Нойхаузера, атаксія Холмса, синдром Олівера — Мак-Фарлана);
— гіпоплазія надниркових залоз у поєднанні з гіпогонадизмом (дефект DAX‑1 гена).
2. Набуті форми (травми, операції, променева терапія, хіміотерапія, інфекції, автоімунні захворювання гіпоталамуса або гіпофіза тощо).
Гіпофізарні гормони (ЛГ, ФСГ) секретуються в пульсуючому режимі 1 раз на 60–90 хв, при цьому ФСГ циркулює в крові довше, ніж ЛГ, тому однократне вимірювання ЛГ і ФСГ не завжди відтворює справжню картину стану гіпоталамо-гіпофізарно-гонадної системи. У хлопчиків після 5–6 міс. життя та дівчаток після 1–1,5 року життя і до періоду статевого дозрівання рутинне дослідження гормонів гіпоталамо-гіпофізарно-гонадної осі для виключення гіпогонадизму не показане, оскільки в цьому періоді настає так звана ювенільна пауза, або фізіологічний гіпогонадизм. Винятком є деякі форми гіпергонадотропного гіпогонадизму (зокрема, синдром Тернера, Клайнфельтера та ін.), при яких не завжди відбувається фізіологічне зниження гонадотропінів до допубертатних показників. Тому найчастіше вторинний (гіпогонадотропний) гіпогонадизм потребує диференціальної діагностики з конституційною затримкою росту й пубертату.
Дослідження антимюллерова гормона (АМГ) як маркера тестикулярної тканини, що нормально функціонує, показане в хлопчиків із підозрою на анорхізм чи крипторхізм, у тому числі для диференціальної діагностики гіпогонадизму (первинний/вторинний). АМГ — це гормон, що синтезується клітинами Сертолі у чоловіків та гранульози примордіальних фолікулів — у жінок. Однією з основних функцій АМГ є забезпечення диференціювання статі в ембріона, а саме пригнічення розвитку жіночих репродуктивних органів із зачатка, що йменується мюллерова протока. У плода жіночої статі синтез АМГ не відбувається, що визначає значну статеву різницю в рівні АМГ при народженні (у хлопчиків рівень АМГ високий, у дівчаток — низький). Крім того, АМГ забезпечує фізіологічний процес опускання яєчок в мошонку, і його недостатній синтез призводить до крипторхізму. Отже, АМГ є незамінним як для диференціальної діагностики крипторхізму та анорхізму, так і для встановлення статі дитини при порушеннях статевого диференціювання [31]. У подальшому рівень АМГ зазнає діаметрально протилежних змін: у пубертатному періоді знижується у хлопчиків і підвищується у дівчаток. У жінок його високий рівень свідчить про наявність склерополікістозу, а також гранульозоклітинного раку яєчників, у той час як в менопаузі його рівень значно знижений.
Найважливішим тестом, крім визначення базального рівня статевих гормонів та АМГ, є проба з гонадотропін-рилізинг гормоном (ГнРГ) [32]. Цей тест слід проводити при досягненні певного віку дитини, зокрема дівчаткам — після 13–13,5 року, хлопчикам — після 13,5–14 років та при досягненні кісткового віку > 10–11 років. Препарати ГнРГ (трипторелін, нафарелін, бусерелін) вводять у дозі 0,1 мг/м2 з оцінкою ЛГ та ФСГ через 30; 60; 90; 120; 240 хв та 24 год після введення препарату. Діагноз вторинного гіпогонадизму виключений при збільшенні ЛГ > 10 МО/л у будь-якій точці. Зазначена проба може бути негативною не тільки при вторинному гіпогонадизмі, але й при конституційній затримці пубертату, тому може знадобитися повторне її проведення через 1–2 роки.
Для більш вірогідної діагностики вторинного гіпогонадизму, крім проби з ГнРГ, слід застосовувати визначення інгібіну В, особливо у хлопчиків. Інгібін В — це глікопротеїн, що секретується клітинами Сертолі яєчка або клітинами гранульози яєчника [31, 33]. Інгібіни вибірково пригнічують секрецію ФСГ (за принципом зворотного зв’язку) [34], регулюючи фолікулогенез та сперматогенез, чинять місцеву паракринну дію в статевих залозах. Синтез інгібіну знижує секрецію гонадоліберину, у той час як андрогени, ФСГ і ІФР‑1 збільшують його секрецію. Тому дефіцит ФСГ асоційований з низьким рівнем інгібіну В у препубертаті [35] та в дорослих чоловіків [31] і може використовуватись для диференціальної діагностики крипторхізму й анорхізму, у діагностиці гіпогонадотропного гіпогонадизму в хлопчиків. Його рівень також знижений в менопаузі у жінок та при порушенні сперматогенезу у чоловіків. Зокрема, за даними Harrington et al., при конституційній затримці росту й пубертату у хлопчиків рівень інгібіну В не був нижчим за 35 нг/мл, на той час як при вторинному гіпогонадизмі його рівень був дуже низьким [36]. Проте в дівчаток використання інгібіну В у диференціальній діагностиці гіпогонадизму й затримки пубертату ще продовжує вивчатися.
Додатково для виключення первинного гіпогонадизму у хлопчиків застосовують пробу з хоріонічним гонадотропіном (ХГ) (триденний тест з 1500 ОД/м2 ХГ) з визначенням вільного тестостерону до проби та на четвертий день після проби.
Для діагностики вторинного гіпогонадизму також застосовують і генетичну діагностику [37] (табл. 4).
Також слід зазначити, що для діагностики, зокрема, вторинного гіпотиреозу та дефіциту пролактину стимуляційні проби не проводять. При вторинному гіпотиреозі, як і при первинному, у сироватці крові вміст вТ4 знижений, але концентрація ТТГ може бути нормальною (у 84 % пацієнтів), низькою або навіть незначно підвищеною [3], у зв’язку з чим при вторинному гіпотиреозі діагностичне значення має лише рівень вТ4. Дефіцит пролактину характерний для пацієнтів з мутаціями PIT1 і PROP1, він не має клінічних проявів і не потребує медикаментозного лікування в дітей та підлітків.
В Україні з 2004 року функціонує реєстр дітей, які отримують гормон росту в зв’язку із гіпофізарним нанізмом, синдромом Тернера, за наявності рідкісної генетичної патології, асоційованої із затримкою росту, при хронічній нирковій недостатності. Він містить інформацію про дітей віком до 17 років включно, а саме: дату народження, дату встановлення діагнозу, антропометричні дані, препарат СТГ, його дозу, тривалість прийому тощо. Згідно з даними реєстру у 2014 р., 1247 дітей отримувало лікування препаратами СТГ, серед яких було зареєстровано 840 дітей з гіпофізарним нанізмом (67,4 %, або поширеність 1 : 9523) і 92 дитини (7,3 %), які мали рідкісну генетичну патологію (зокрема, синдром Рассела — Сільвера, синдром Секкеля, Лоуренса — Муна — Барде — Бідля, Прадера — Віллі, Корнеллі де Ланге, Аарського, пікнодизостоз тощо). Інші діти отримували препарати СТГ у зв’язку із синдромом Тернера (24,7 %) та хронічною нирковою недостатністю (0,6 %). Отже, у зв’язку зі значною поширеністю гіпофізарного нанізму в Україні проблема його адекватної діагностики є вкрай важливою.
Таким чином, впровадження в рутинну практику дитячого ендокринолога двох стимуляційних тестів з визначенням СТГ у багатьох часових точках, оцінка ІФР‑1 за стандартним відхиленням, розширення показань для використання АМГ та інгібіну В (у хлопчиків), коректне проведення проби з ГнРГ, а також використання генетичних методів дозволить значно покращити діагностику гіпопітуїтаризму в дітей, у тому числі уникнути хибного діагнозу й подальшого необґрунтованого лікування.
Тести, рекомендовані «Сінево»
1268 Пакет № 47 (фактори росту) (соматотропний гормон (гормон росту, СТГ); інсуліноподібний фактор росту 1 (ІФР‑1, ІGF‑1); інсуліноподібний фактор росту зв’язуючого білка 3 (IGF-BP3)).
Список литературы
1. Murray P.G., Dattani M.T., Clayton P.E. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence // Arch. Dis. Child. — 2016. — Vol. 101. — Р. 96-100.
2. Reh C.S., Geffner M. Somatotropin in the treatment of growth hormone deficiency and Turner syndrome in pediatric patients: a review // Clinical Pharmacology: Advances and Applications. — 2010. — Vol. 2. — Р. 111-122.
3. Федеральные клинические рекомендации по диагностике и лечению гипопитуитаризма у детей и подростков / Российское общество эндокринологов. — 2013. — 43 с.
4. Kuczmarski R.J., Ogden C.L., Grummer-Strawn L.M. et al. CDC growth charts: United States // Vital and Health Statistics. — 2000. — Vol. 314. — Р. 1-27.
5. Greulich W., Pyle S. Radiographic Atlas of Skeletal Development of the Hand and Wrist. — Stanford: Stanford University Press, 1959.
6. Ergun-Longmire B., Wajnrajch M. Growth and Growth Disorders. — 2013.
7. Rakover Y., Lavi I., Masalah R. et al. Comparison between four immunoassays for growth hormone (GH) measurement as guides to clinical decisions following GH provocative tests // J. Pediatr. Endocrinol. Metab. — 2000. — Vol. 13. — Р. 637-43.
8. Arsene C.G., Kratzsch J., Henrion A. Mass spectrometry — an alternative in growth hormone measurement // Bioanalysis. — 2014. — Vol. 6. — Р. 2391-402.
9. Lazar L., Phillip M. Is sex hormone priming in peripubertal children prior to growth hormone stimulation tests still appropriate? // Horm. Res Paediatr. — 2010. — Vol. 73(4). — Р. 299-302.
10. Marin G., Domene H.M., Barnes K.M. et al. The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys // J. Clin. Endocrinol. Metab. — 1994. — Vol. 79. — Р. 537-41.
11. Consensus Guidelines for the Diagnosis and Treatment of Growth Hormone (GH) Deficiency in Childhood and Adolescence: Summary Statement of the GH Research Society // The Journal of Clinical Endocrinology and Metabolism. — 2000. — Vol. 85(11). — Р. 3990-93.
12. Абатуров О.Є., Агафонова О.О., Герасименко О.М. Захворювання гіпофізарно-надниркової системи у дітей. — 2013. — 206 с.
13. Cogan J.D., Wu W., Phillips J.A. et al. The PROP12-base pair deletion is a common cause of combined pituitary hormone deficiency // J. Clin. Endocrinol. Metab. — 1998. — Vol. 83. — Р. 3346-3349.
14. Rodriguez R., Andersen B. Cellular determination in the anterior pituitary gland: PIT‑1 and PROP‑1 mutations as causes of human combined pituitary hormone deficiency // Minerva Endocrinol. — 2003. — Vol. 28. — Р. 123-133.
15. De Graaff L.C., Argente J., Veenma D.C. et al. PROP1, HESX1, POU1F1, LHX3 and LHX4 mutation and deletion screening and GH1 P89L and IVS3+1/+2 mutation screening in a Dutch nationwide cohort of patients with combined pituitary hormone deficiency // Horm. Res Paediatr. — 2010. — Vol. 73. — Р. 363-371.
16. Böttner A., Keller E., Kratzsch J. et al. PROP1mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis // J. Clin. Endocrinol. Metab. — 2004. — Vol. 89. — Р. 5256-5265.
17. Mendonca B.B., Osorio M.G., Latronico A.C. et al. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301, G302 in the PROP1 gene // J. Clin. Endocrinol. Metab. — 1999. — Vol. 84. — Р. 942-945.
18. Wu W., Cogan J.D., Pfaffle R.W. et al. Mutations in PROP1 cause familial pituitary hormone deficiency // Nat. Genet. — 1998. — Vol. 18. — Р. 147-149.
19. Fofanova O., Takamura N., Kinoshita E., Yamashita S. et al. Compound heterozygous deletion of the PROP‑1 gene in children with combined pituitary hormone deficiency // J. Clin. Endocrinol. Metab. — 1998. — Vol. 83. — Р. 2601-2604.
20. Lemos M.C., Gomes L., Bastos M. et al. PROP1gene analysis in Portuguese patients with combined pituitary hormone deficiency // Clin. Endocrinol. — 2006. — Vol. 65. — Р. 479-485.
21. Lebl J., Vosahlo J., Pfaeffle R.W. et al. Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects // Eur. J. Endocrinol. — 2005. — Vol. 153. — Р. 389-396.
22. Brook C.G.D., Clayton P.E., Brown R.S. Brook’s clinical pediatric endocrinology. — 6th ed. — Singapore, Hong Kong: John Wiley, Sons, Ltd, 2009. — Р. 76.
23. Dattani M.T., Robinson I.C. The molecular basis for developmental disorders of the pituitary gland in man // Clin. Genetics. — 2000. — Vol. 57. — Р. 337-346.
24. Agarwal G., Bhatia V., Cook S., Thomas P.Q. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion // J. Clin. Endocrinol. Metab. — 2000. — Vol. 85. — Р. 4556-4561.
25. Dattani M.T. GH deficiency might be associated with normal height in PROP1 deficiency // Clin. Endocrinol. — 2002. — Vol. 57. — Р. 157-158.
26. Cushman L.J., Showalter A.D., Rhodes S.J. Genetic defects in the development and function of the anterior pituitary gland // Annals of Medicine. — 2002. — Vol. 34. — Р. 179-191.
27. Mody S., Brown M.R., Parks J.S. The spectrum of hypopituitarism caused by PROP1 mutations. Best Practice // Research Clin. Endocrinol. Metab. 2002. — Vol.16. — Р. 421-431.
28. Navardauskaite R., Dusatkova P., Obermannova B. et al. High prevalence of PROP1 defects in Lithuania: phenotypic findings in an ethnically homogenous cohort of patients with multiple pituitary hormone deficiency // J. Clin. Endocrin. Metab. First published ahead of print October 31, 2013 as doi:10.1210/jc.2013-3090.
29. Bonomi M., Libri D.V., Guizzardiet F. et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism // Asian Journal of Andrology. — 2012. — Vol. 14(1). — Р. 49-56.
30. Федеральные клинические рекомендации (протоколы) по диагностике и лечению гипогонадизма у детей / Российское общество эндокринологов. — М., 2013. — 22 с.
31. Li H.W., Anderson R.A., Yeung W.S. et al. Evaluation of serum antimullerian hormone and inhibin B concentrations in the differential diagnosis of secondary oligoamenorrhea // Fertil. Steril. — 2011. — Vol. 96. — Р. 774-779.
32. Harrington J., Palmert M.R. Distinguishing Constitutional Delay of Growth and Puberty from Isolated Hypogonadotropic Hypogonadism: Critical Appraisal of Available Diagnostic Tests // J. Clin. Endocrinol Metab. — 2012. — Vol. 97(9). — Р. 3056-3067.
33. Raivio T., Dunkel L. Inhibins in childhood and puberty // Best Pract. Res. Clin. Endocrinol. Metab. — 2002. — Vol. 16. — Р. 43-52.
34. Anderson R.A., Sharpe R.M. Regulation of inhibin production in the human male and its clinical applications // Int. J. Androl. — 2000. — Vol. 23. — Р. 136-144.
35. Raivio T., Saukkonen S., Jaaskelainen J. et al. Signaling between the pituitary gland and the testes: inverse relationship between serum FSH and inhibin B concentrations in boys in early puberty // Eur. J. Endocrinol. — 2000. — Vol. 142. — Р. 150-156.
36. Harrington J., Palmert M.R. Distinguishing Constitutional Delay of Growth and Puberty from Isolated Hypogonadotropic Hypogonadism: Critical Appraisal of Available Diagnostic Tests // J. Clin. Endocrinol. Metab. — 2012. — Vol. 97(9). — P. 3056-3067.
37. Bianco Suzy D.C., Kaiser U.B. The genetic and molecular basis of idiopathic hypogonadotropic Hypogonadism // Nat. Rev. Endocrinol. — 2009. — Vol. 5(10). — Р. 569-576.

/65.jpg)
/67.jpg)