Резюме
Метою наведеного огляду був аналіз даних літератури щодо молекулярно-генетичної основи та діагностики ендокринної патології. Проведено огляд наукової літератури за ключовими словами «гени», «ендокринні хвороби», «молекулярна діагностика», «прогормони», «ядерні рецептори і фактори транскрипції» з використанням пошукової системи PubMed. Беручи до уваги дослідження, проведені в останні 10 років, проаналізовані тези 144 статей. Більш детально вивчено результати дослідження, висвітлені у 32 статтях. Серед наведених нозологій — різні спадкові форми гіпопітуїтаризму, порушення біосинтезу стероїдних гормонів, порушення формування статі, моногенні форми цукрового діабету (ЦД) та інші захворювання. Виділено патологію, що пов’язана з мутацією генів, які кодують білкові прогормони, рецептори, ферменти біосинтезу стероїдів, молекули внутрішньоклітинного сигналінгу, транспортні білки, іонні канали, фактори транскрипції. Серед ендокринних захворювань, асоційованих із дефектами генів, які кодують білкові прогормони, найбільш поширеними є дефекти гена GH1; серед захворювань, асоційованих із дефектами генів, які кодують ферменти, найбільш поширеними є дефекти гена CYP21A2 (21-гідроксилази). Частіше мутації генів, які кодують білки, що відносяться до класу рецепторів, пов’язані із G-білком (G-protein coupled receptors). Більшість мутацій, пов’язаних із МЕН-2А, зосереджено в багатому цистеїном регіоні рецептора Ret. Відомо більше 70 моногенних синдромів, при яких відзначається клінічно виражена толерантність до глюкози і діагностується та чи інша форма ЦД, також виявлений ЦД, зумовлений мутацією мітохондріального гена (мутація tRNALeu, UUR). Самою частою з усіх моногенних форм є ожиріння, спричинене мутацією гена рецептора меланокортину 4-го типу (MC4R). У хворих на вроджений гіпотиреоз підвищена частота алелей HLA (Bw44, Aw24, B18) та описані мутації в генах TITF1, TITF2, FOXE1, PAX8 (транскрипційні маркери нормального онтогенезу щитоподібної залози).
Целью представленного обзора был анализ данных литературы относительно молекулярно-генетической основы и диагностики эндокринной патологии. Проведен обзор научной литературы по ключевым словам «гены», «эндокринные болезни», «молекулярная диагностика», «прогормоны», «ядерные рецепторы и факторы транскрипции» с использованием поисковой системы PubMed. Принимая во внимание исследования, проведенные в последние 10 лет, проанализированы результаты 144 статей. Более подробно изучены результаты исследования, которые освещены в 32 статьях. Среди представленных нозологий — различные наследственные формы гипопитуитаризма, нарушения биосинтеза стероидных гормонов, нарушение формирования пола, моногенные формы сахарного диабета и другие заболевания. Выделена патология, которая связана с мутацией генов, кодирующих белковые прогормоны, рецепторы, ферменты биосинтеза стероидов, молекулы внутриклеточного сигналинга, транспортные белки, ионные каналы, факторы транскрипции. Среди эндокринных заболеваний, ассоциированных с дефектами генов, кодирующих белковые прогормоны, наиболее распространенными являются дефекты гена GH1; среди заболеваний, ассоциированных с дефектами генов, кодирующих ферменты, — дефекты гена CYP21A2 (21-гидроксилазы). Чаще мутации генов, кодирующих белки, которые относятся к классу рецепторов, связаны с G-белком (G-protein coupled receptors). Большинство мутаций, связанных с МЭН-2А, сосредоточены в богатом цистеином регионе рецептора Ret. Известно более 70 моногенных синдромов, при которых отмечается клинически выраженная толерантность к глюкозе и диагностируется та или иная форма сахарного диабета, также обнаружен сахарный диабет, обусловленный мутацией митохондриального гена (мутация tRNALeu, UUR). Из всех моногенных форм ожирения наиболее часто встречается ожирение, вызванное мутацией гена рецептора меланокортин 4-го типа (MC4R). У больных с врожденным гипотиреозом высокая частота аллелей HLA (Bw44, Aw24, B18) и описаны мутации в генах TITF1, TITF2, FOXE1, PAX8 (транскрипционные маркеры нормального онтогенеза щитовидной железы).
The purpose of the review was analysis of literature data relating to the molecular genetic basis and diagnosis of endocrine pathology. We searched for published and unpublished researches using Pubmed as the search engine by the keywords: ‘genes’, ‘endocrine diseases’, ‘molecular diagnostics’, ‘prohormones’, ‘nuclear receptors and transcription factors’, taking into consideration studies conducted over the last 10 years, citation review of relevant primary and review articles, conference abstracts, personal files, and contact with expert informants. The criterion for the selection of articles for the study was based on their close relevance to the topic, thus out of 144 analyzed articles, the findings of the researchers covered in 32 articles were crucial. The described nosologies presented various hereditary forms of hypopituitarism, disturbances of steroid hormone biosynthesis, abnormal gender formation, monogenic forms of diabetes mellitus, endocrine tumors, etc. Pathology is identified that is associated with a mutation of genes encoding protein prohormones, receptors, steroid biosynthesis enzymes, intracellular signaling molecules, transport proteins, ion channels, and transcription factors. Among the endocrine diseases associated with defects in genes encoding protein prohormones, the defects of the GH1 gene are most common, the defects in the gene CYP21A2 (21-hydroxylase) are among diseases associated with defects in genes encoding enzymes. More often mutations of genes encoding proteins belong to the class of G-protein coupled receptors. Most of the mutations associated with MEN-2A are concentrated in the rich cysteine region of the Ret receptor. More than 70 monogenic syndromes are known, in which there is a marked tolerance to glucose and some form of diabetes mellitus is diagnosed, diabetes mellitus caused by mutation of the mitochondrial gene (mutation tRNALeu, UUR) is also detected. Of all the monogenic forms of obesity, the most common is obesity caused by mutation of melanocortin 4 receptor gene (MC4R). In patients with congenital hypothyroidism, the high frequency of HLA alleles (Bw44, Aw24, B18) and mutations in the genes TITF1, TITF2, FOXE1, PAX8 (transcription markers of normal thyroid ontogenesis of the thyroid gland) are described.
Останніми роками завдяки успіхам молекулярної генетики, до яких, безумовно, слід віднести і розшифровку генома людини, відзначений колосальний прогрес медичної науки в цілому, особливо ендокринології. Впровадження в фундаментальну і клінічну медицину сучасних досягнень генетики та молекулярної біології істотно змінило наші уявлення про етіопатогенез і, як наслідок, можливості діагностики, лікування і профілактики багатьох захворювань.
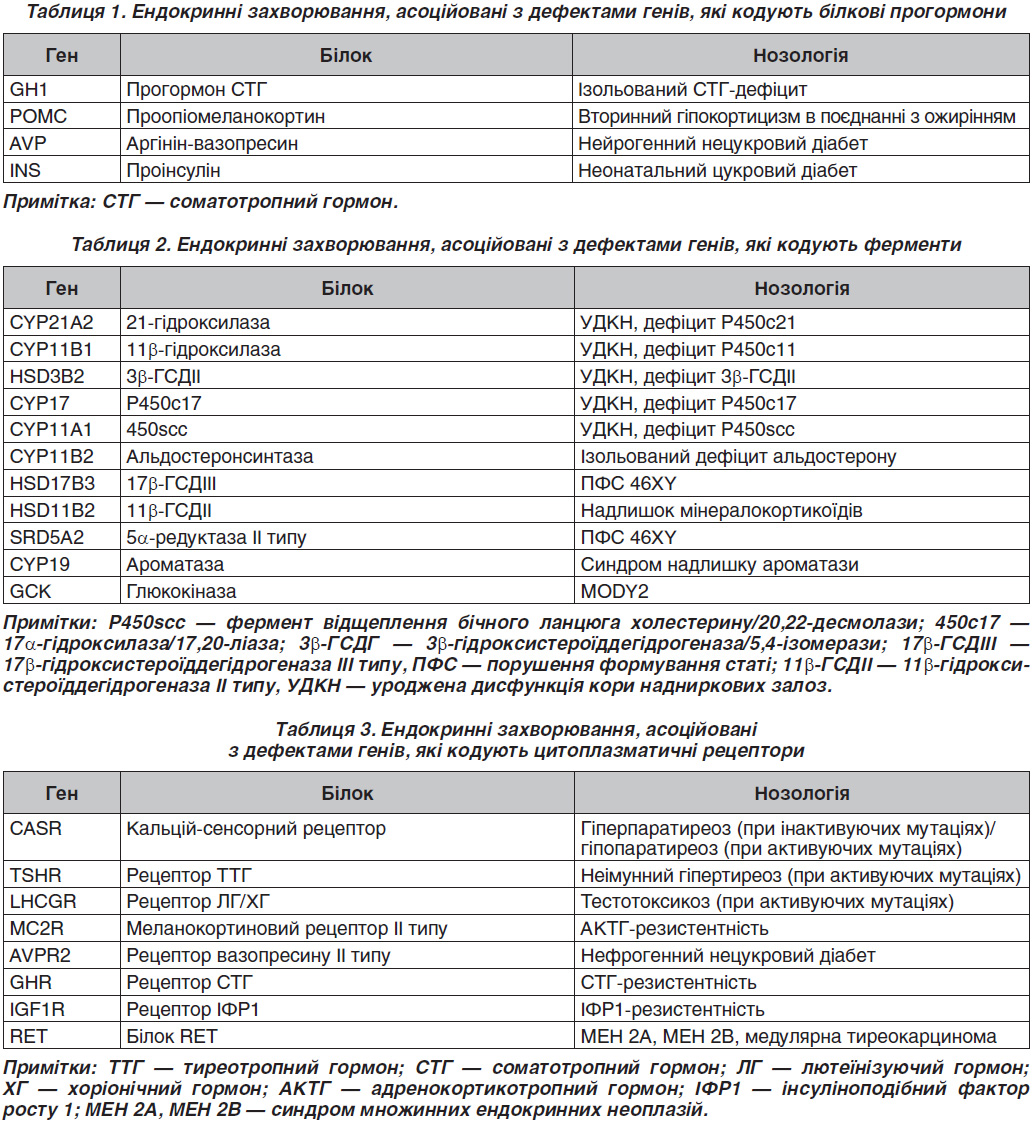
Багато ендокринних захворювань мають спадкову природу та асоціюються з дефектом якогось певного гена, зокрема виділяють патологію, пов’язану з мутацією генів, які кодують білкові прогормони, рецептори, ферменти біосинтезу стероїдів, молекули внутрішньоклітинного сигналінгу, транспортні білки, іонні канали, фактори транскрипції тощо (табл. 1–5) [1].
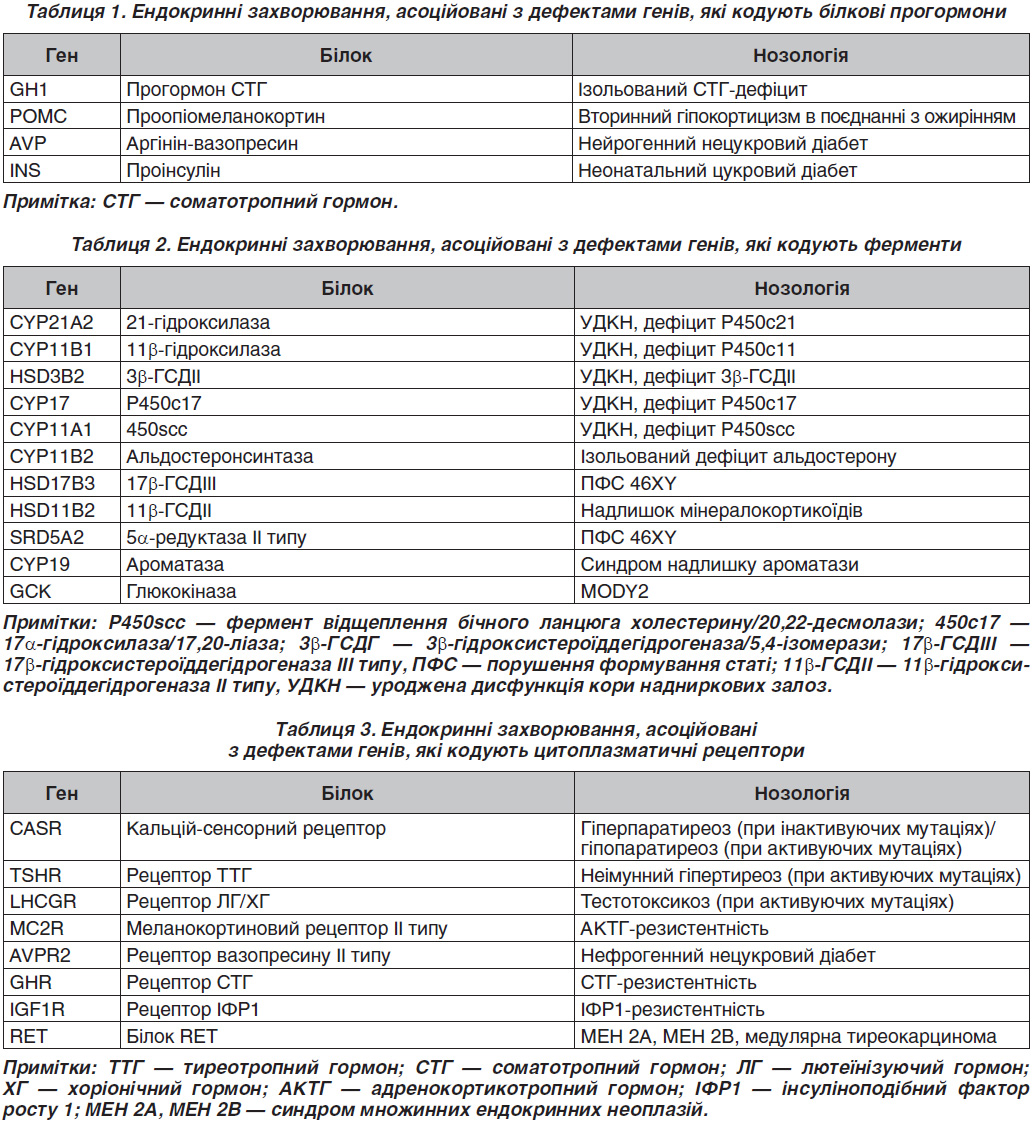
Серед ендокринних захворювань, асоційованих із дефектами генів, що кодують білкові прогормони, найбільш поширеними є дефекти гена GH1, який кодує прогормон соматотропного гормона (СТГ) [2]. Фенотипово захворювання характеризується вираженою низькорослістю, що зумовлено ізольованим дефіцитом СТГ. Залежно від типу успадкування та вираженості СТГ-дефіциту виділяють дві аутосомно-рецесивні форми (тип IA і IB) і аутосомно-домінантну форму (тип II). У російській популяції найбільш поширеним є тип II, більшість випадків якого було асоційовано зі сплайсинг-мутаціями в гені GH1 [1]. Аутосомно-рецесивні форми ізольованого СТГ-дефіциту трапляються рідше, між тим важливо пам’ятати про необхідність правильної діагностики типу IA. В літературі описано пацієнта з делецією гена GH1, тому що для цієї форми характерна резистентність до екзогенного гормона росту, і ростостимулюючий ефект можливий тільки при використанні рекомбінантного ІФР1 [3].
Дефект гена POMC (раніше неописана гомозиготна мутація W58X) був виявлений у дитини з ранньої маніфестацією ожиріння, гіпоглікемічним синдромом і рудим кольором волосся [4]. У сім’ї, в якій у декількох поколіннях відзначалася клініка нейрогенного (центрального) нецукрового діабету, встановлена гетерозиготна мутація в гені AVP (c. 230-232delAGG p.E78del) [1].
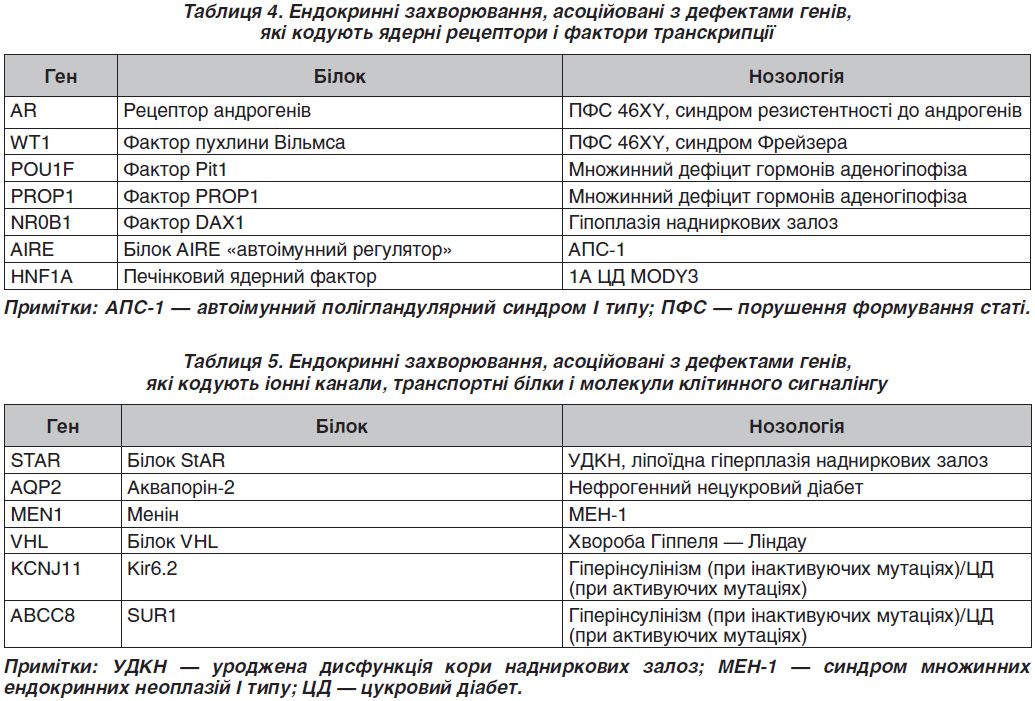
Серед захворювань, асоційованих із дефектами генів, що кодують ферменти, найбільш поширеними є дефекти гена CYP21A2 (21-гідроксилази). Уроджена дисфункція кори надниркових залоз (УДКН) є найбільш частою причиною гіперандрогенії надниркового генезу. УДКН — аутосомно-рецесивне захворювання, зумовлене у 95 % випадків дефіцитом ферменту 21-гідроксилази. На даний час прийнято виділяти 6 варіантів УДКН: 1) дефіцит протеїну (ліпоїдна гіперплазія надниркових залоз, синдром Прадера); 2) дефіцит 3-стероїддегідрогенази; 3) дефіцит 17α-гідроксилази Л7, 20-ліази [Р450с17] (синдром Беглієрі); 4) дефіцит 21-гідроксилази [Р450с21]; 5) дефіцит 11-гідроксилази [Р450с11]; 6) дефіцит Р450-оксидоредуктаз.
У європейській популяції частота УДКН досить висока і становить 1 : 2,5–10 000 новонароджених. Загальнопопуляційна частота перебуває в межах від 0,3 до 1 % [5]. Некласична УДКН трапляється при недостатності 21-гідроксилази (Р450с21), 11-гідроксилази (Р450с11в), 3-гідроксистероїдної дегідрогенази (3-ГСД) і являє собою м’який варіант 21-гідроксилазного дефіциту. У людини виявлено два гени, що кодують послідовність 21-гідроксилази, — CYP21B і CYP21A [6]. Ці гени гомологічні, але транскрипційно активний тільки один із них — CYP21B. Ген CYP21A містить кілька мутацій, через які його експресія неможлива. CYP21B і CYP21A розташовуються поруч із двома генами четвертого компонента комплементу (С4а, С4В). У хворих із недостатністю 21-гідроксилази знайдені заміни, делеції і точкові мутації в гені CYP21B. Приблизно у 70–75 % хворих із гаплотипами HLA високого ризику виявляється нормальний ген CYP21B [7]. Мабуть, у таких хворих існують точкові мутації гена CYP21B, які неможливо виявити простими методами гібридизації ДНК. У дітей із вірильною формою УДКН часто виявляються мутації, при яких зберігається можливість синтезу в надниркових залозах невеликої кількості альдостерону, тому клінічних проявів втрати солі у них немає [8].
Єдиним відомим на сьогодні спадковим захворюванням, в основі якого лежить підвищення активності стероїдогенних ферментів, є синдром надлишку ароматази [9]. Клінічно даний стан проявляється допубертатною гінекомастією у чоловіків і раннім менархе і макромастією у жінок.
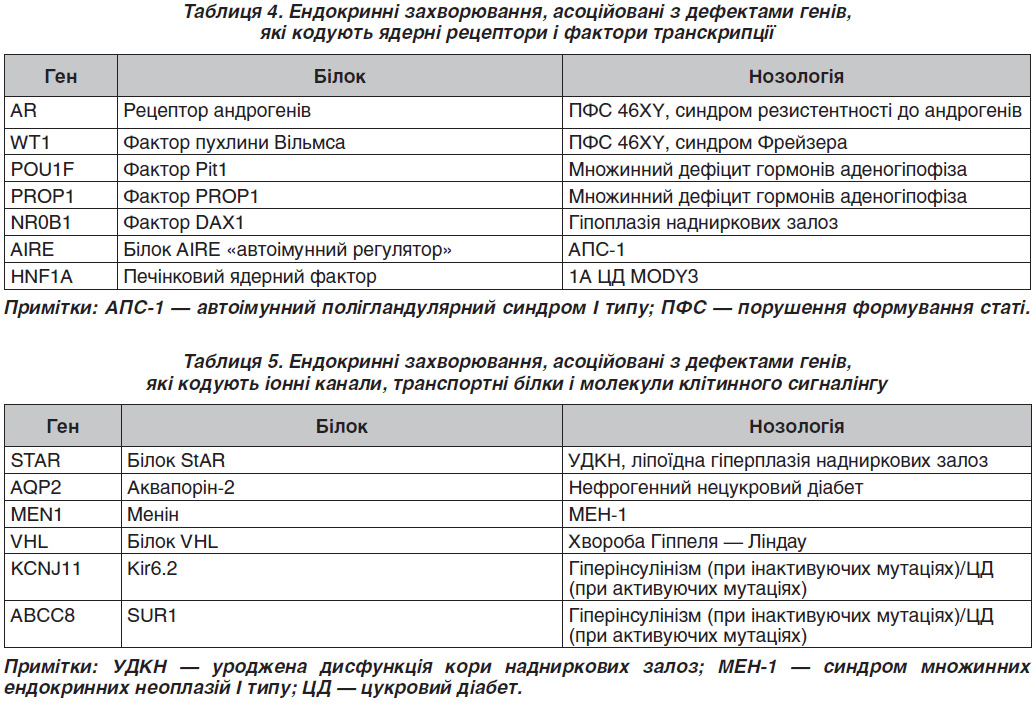
Перші п’ять із наведених у табл. 3 генів, які кодують білки, відносяться до класу рецепторів, пов’язаних із G-білком (G-protein coupled receptors) [10, 11]. Унікальною особливістю даних рецепторів є те, що їх зміни залежно від локалізації й характеру заміни амінокислот можуть призводити як до зниження, так і до підвищення їх функції. У нормі активація кальцій-сенсорного рецептора (CASR) під впливом іонів кальцію призводить до зниження функції паратиреоцитів. CASR є G-білком, який експресується в паратиреоїдний гормон, що продукується головними клітинами прищитоподібної залози і клітин, що вистилають канальці нирок [12]. У силу своєї здатності відчувати невеликі зміни в концентрації кальцію CASR відіграє істотну роль у підтримці мінерального іонного гомеостазу. Інактивуюча складова — гетерозиготна мутація в гені CASR (c.1683delAp.S564fsX636/C749) — була виявлена у дитини з тяжким уродженим гіперпаратиреозом [13–15].
З 1993 року, коли була виявлена мутація в протоонкогені Ret у родинах із МЕН-2А, МЕН-2В, виникла можливість ідентифікувати пацієнтів, які мають ризик розвитку раку щитоподібної залози (ЩЗ). Вважають, що синдроми МЕН зумовлені мутаціями в клітинах нервового гребінця. Причиною синдрому МЕН-1
служать мутації гена-супресора пухлинного росту (11q13) [16]. Цей ген кодує білок менін, який регулює проліферацію клітин — похідних нервового гребінця. Розвиток синдромів МЕН-2 пов’язаний із виникненням мутацій у RET-протоонкогені, який локалізується на довгому плечі 10-ї хромосоми. Більшість мутацій, пов’язаних із МЕН-2А, зосереджені в багатому цистеїном регіоні рецептора Ret. Пацієнти з МЕН-2В мають одиничну крапкову мутацію в кодоні 918 Ret. Пряме тестування ДНК — це високоточна методика виявлення родичів, які успадкували специфічну мутацію, пов’язану з МЕН-2А та МЕН-2В. У випадках, коли виявлені зміни в геномі, характерні для МЕН-2 синдромів, ситуація доволі однозначна: гіперплазія С-клітин або ж медулярна карцинома ЩЗ виявлена у всіх носіїв гена, навіть у наймолодших. Особливо важливим є своєчасне (доклінічне) виявлення пацієнтів із МЕН-2В синдромом, тому що серед тих, кому діагноз було встановлено вже за наявності клінічних ознак, практично половина вмирає від цього захворювання, а інші страждають від тяжких проявів хвороби. У сім’ях із МЕН-2В скринінг має відбутися протягом першого року життя і повинен включати пошук характерного фенотипу [17].
Мутації в гені AR асоційовані з однією з найбільш частих причин порушення формування статі (ПФС) 46XY — синдрому резистентності до андрогенів (тестикулярна фемінізація) [18]. На відміну від інших форм ПФС 46XY при цьому захворюванні відсутній ефект від екзогенних андрогенів, тому в більшості випадків рекомендується адаптація в жіночій статі [19].
Гіпофізарний фактор транскрипції (PIT-1) бере участь у регуляції експресії генів пролактину, соматотропіну і тиреотропного гормона, а також впливає на диференціацію і проліферацію клітин гіпофіза [20]. Мутації в гені PIT-1 призводять до гіпоплазії гіпофіза, синдрому уродженого гіпотиреозу. PIT-1 є членом POU-домену, в який входить група транс–крипційних регуляторів, що відіграють важливу роль у диференціації і проліферації клітин.
PIT-1 має три варіанти сплайсингу. Основний тип — Pit1-α, два інших — Pit1-β і Pit-1T. Усі варіанти сплайсингу є біологічно активними. На даний час виявлено кілька варіантів гена PIT-1, що визначаються точковими мутаціями. Три мутації — Pit1I3H (C- і D-алелі), Pit1I3N (M- і N-алелі) і Pit1I3NL (G- і H-алелі) локалізовані в третьому інтроні. Ці мутації виявляються за допомогою рестриктаз HinfI, NciI іNlaIII відповідно. Так само по одній мутації Pit1I4N (E- і F-алелі), Pit1I5, Pit1E6H (A- і B-алелі) були виявлені в четвертому, п’ятому інтроні і шостому екзоні гена PIT-1.
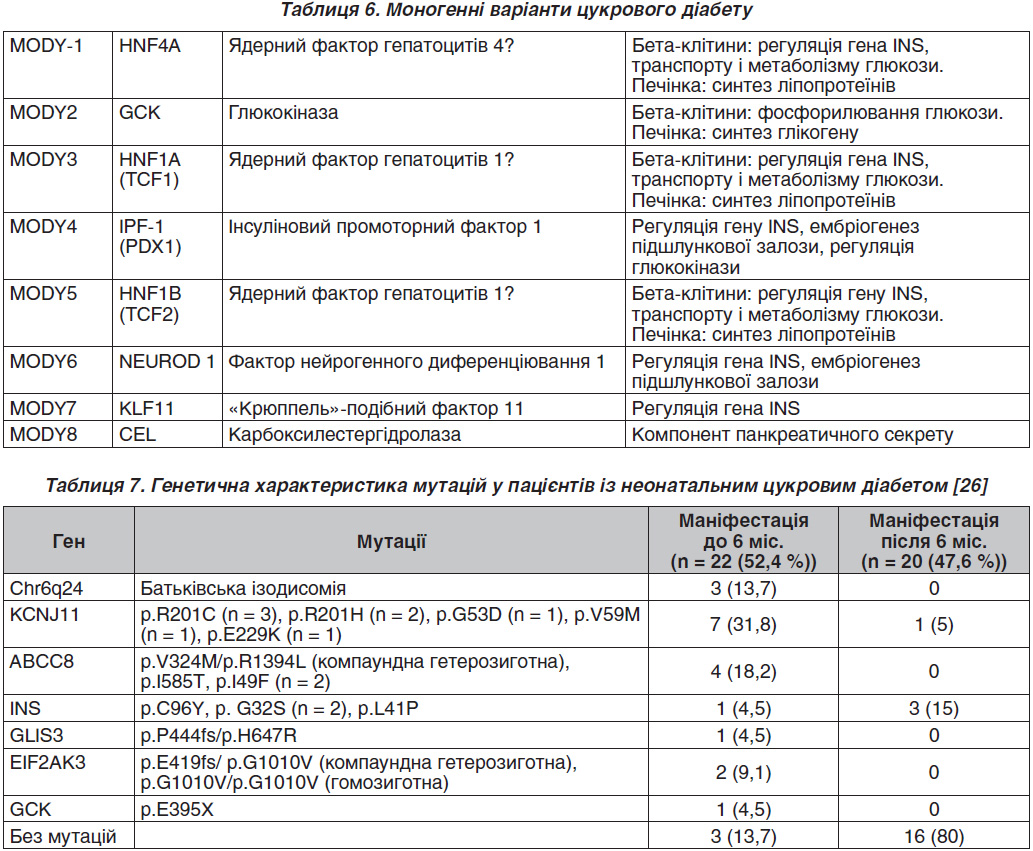
На даний час відомо більше 70 моногенних синдромів, при яких відзначається клінічно виражена толерантність до глюкози і діагностується та чи інша форма ЦД [21]. При цьому на частку всіх рідкісних моногенних форм ЦД доводиться не більше 1 % таких випадків, тоді як переважна більшість ЦД представлена типовими мультифакторними захворюваннями, частка спадкового компонента яких перевищує 50 % [22]. Про полігенну природу ЦД свідчить порівняно низький рівень сімейного ризику хвороби. Його мультифакторну природу доводить наявність багатьох досить добре відомих несприятливих зовнішніх чинників, що на тлі несприятливого генотипу призводять до розвитку захворювання. Гени схильності до ЦД значно поширені в популяціях, але при цьому кожен із них сам по собі, як правило, недостатній для розвитку ЦД. Тільки наявність певних комбінацій несприятливих алельних варіантів таких генів на тлі провокуючих зовнішніх факторів середовища може призводити до розвитку ЦД того чи іншого типу [23]. До цієї групи належать також типи ЦД, в патогенезі яких чітко встановлена причина захворювання, зумовлена порушенням виділених генів. Розвиток ЦД в таких випадках поєднується з моногенним дефектом функції β-клітин (табл. 6).
Важливим відкриттям останніх років є також виявлення ЦД, зумовленого мутацією мітохондріального гена, зокрема мутацією tRNALeu (UUR). Уперше ця точкова мутація була описана при MELAS-синдромі (мітохондріальна міопатія, лактат-ацидоз, енцефалопатія і інсультоподібні епізоди). Одним із компонентів MELAS-синдрому є наявність ЦД із втратою або без втрати слуху [24]. До інших форм ЦД належать сімейні випадки, що перебігають із клінічною картиною ЦД2, зумовлені мутантними або аномальними формами інсуліну («чиказький інсулін», «лос-анджелеський інсулін», «інсулін Вакаяма»), які зберігають лише 5–10 % біологічної активності нативного інсуліну. Розвиток ЦД може бути зумовлений генетичними порушеннями, пов’язаними з периферичною дією інсуліну, тобто бути наслідком мутацій гена рецепторів інсуліну [25].
До генетичних синдромів, асоційованих із ЦД, відносяться:
— синдром Вольфрама, DIDMOAD-синдром, АР (цукровий діабет, нецукровий діабет, АЗН, ней–росенсорна туговухість). Мутація гена WFS1, що кодує білок вольфрамін;
— синдром Рабсона — Менденхолла (дисплазія ясен і зубів, швидкий ріст нігтів і гірсутизм);
— синдром Донохью (тяжка затримка внутрішньоутробного розвитку, повна відсутність підшкірної жирової клітковини при народженні, лицевий дисморфізм («ельфоподібне» обличчя з великими кулястими очима, відстовбурченими вухами і мікрогнатія), роздутий живіт і порушення гомеостазу глюкози (гіпоглікемія натще і гіперглікемія після їжі). Зазвичай хворі гинуть в перші 2 роки життя;
— синдром Альстрема (дистрофія сітківки, ней–росенсорна туговухість, раннє ожиріння (у віці до 1 року), цукровий діабет, гіперліпідемія, кардіоміопатія, патологія печінки);
— синдром Роджера (мегалобластна анемія, чутлива до тіаміну, мутація гена SLC19A2);
— мітохондріальний ЦД.
Для багатьох генів-кандидатів ЦД 1-го типу добре встановлений поліморфізм, чітко ідентифіковані несприятливі алелі. У генної мережі виділяють наступні основні групи генів:
— гени головного комплексу гістосумісності (HLA), відповідальні за продукцію, транспорт і подання на клітинних мембранах відповідних антигенів (HLA, клас II та III);
— гени, які контролюють продукцію цитокінів (IL1, IL1R1, IL1N1, TNF-α); ген інсуліну INS;
— гени, що включають механізми деструкції, захисту і репарації β-клітин острівців Лангерганса підшлункової залози (SOD2, HSP-70, NOS2).
Провідна роль у загибелі β-клітин, що продукують інсулін, відводиться генам HLA класу II — DRB1, DQА1 і DQB (гаплотипи DR3 і DR4), вклад яких у розвиток ЦД 1-го типу в різних популяціях, за узагальненими даними, перебуває в межах від 30 до 40 %.
Дані про неонатальний цукровий діабет наведені у табл. 7.
За даними [27], захворюваність на неонатальний цукровий діабет (НЦД) становить 1 на 103 460 дитячого населення віком до 18 років. Із 42 обстежених дітей, які захворіли у віці до 9 місяців, генетичну природу НЦД було підтверджено у 23 пацієнтів (54,8 %), серед них було 7 пацієнтів із транзиторним НЦД (30,4 %) і 16 (69,6 %) — з перманентним.
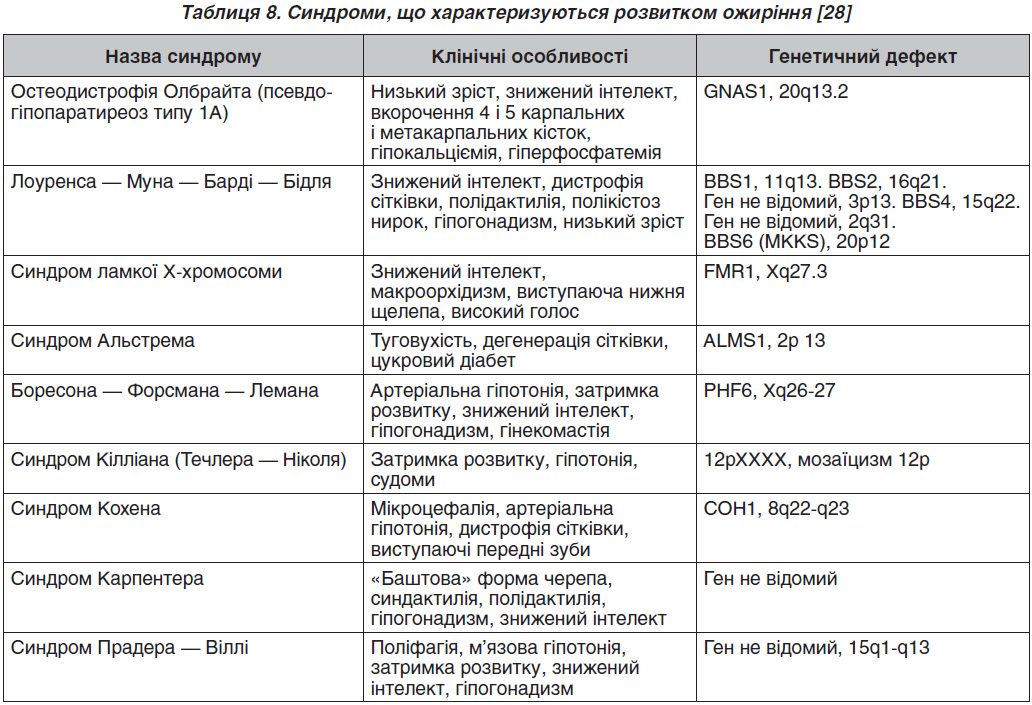
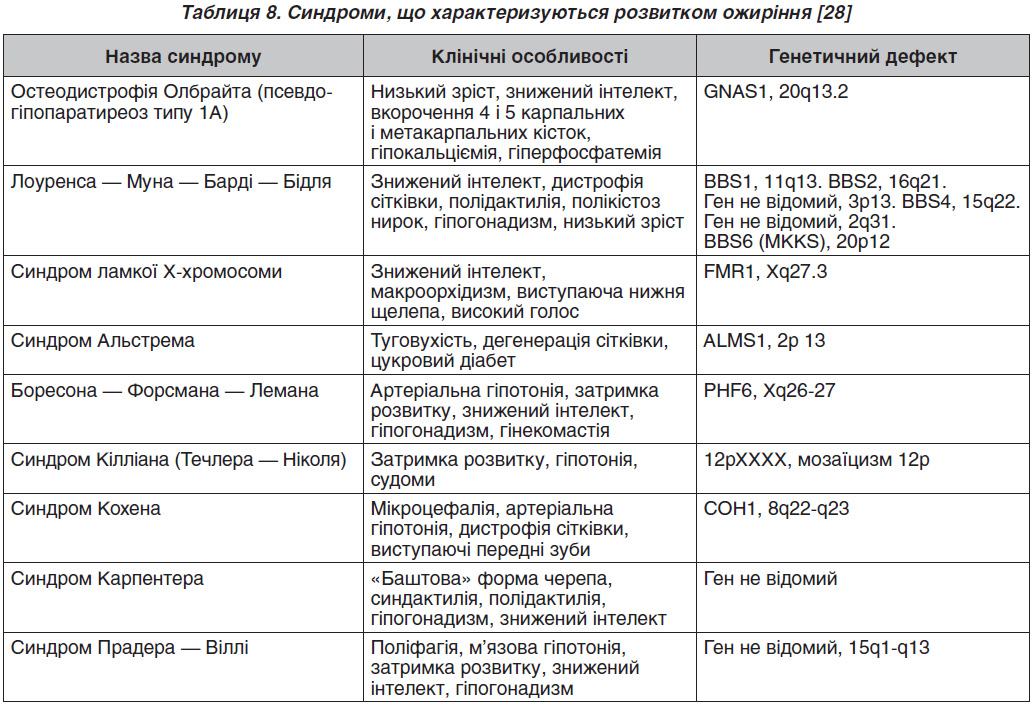
У табл. 8 наведені синдроми, що асоціюються з розвитком ожиріння.
Молекулярно-генетичне дослідження (визначення каріотипу, мутацій певних генів) проводиться при підозрі на моногенне ожиріння і синдромальні форми. Особливістю синдромальних форм ожиріння є наявність вираженої неврологічної симптоматики — затримка психомоторного розвитку, знижений інтелект. У даному випадку бажана консультація генетика, проведення генетичних досліджень з урахуванням клінічної картини і фенотипічних особливостей. Моногенні форми ожиріння відрізняються раннім дебютом (з перших місяців життя), поліфагією з розвитком вираженого, нерідко морбідного ожиріння до 3–5 років. Слід зазначити, що найчастішою з усіх моногенних форм є ожиріння, спричинене мутацією гена рецептора меланокортину 4-го типу (MC4R), що клінічно характеризується поєднанням раннього морбідного ожиріння на тлі поліфагії і високорослості [29]. У зв’язку з цим при наявності у дитини до трьох років вираженого ожиріння показано дослідження гена MC4R. Однак необхідно відзначити, що час–тота народження навіть цієї «найчастішої» форми становить не більше 0,5–4 %.
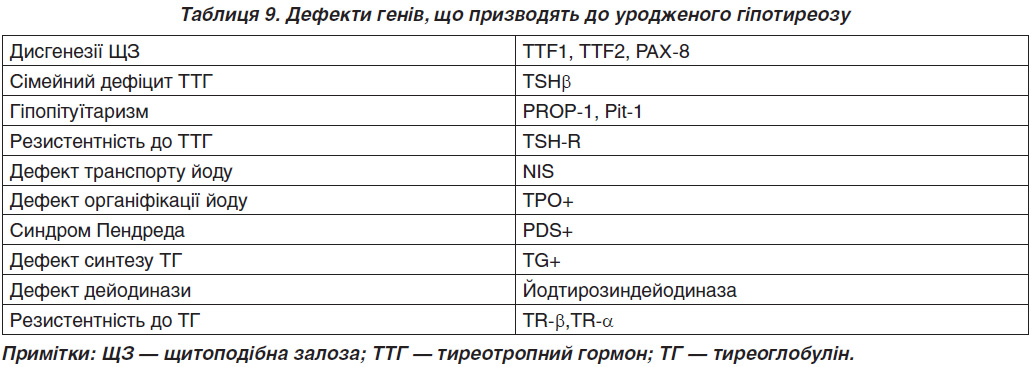
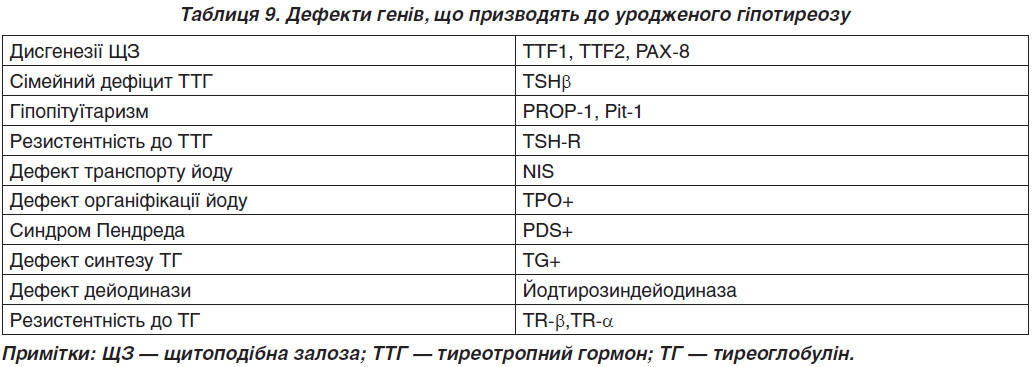
Приблизно 2 % дисгенезу ЩЗ — це сімейна форма хвороби [30]. На сьогодні описані мутації в генах TITF1, TITF2, FOXE1, PAX8 (транскрипційні маркери нормального онтогенезу ЩЗ, табл. 9) [31, 32]. У хворих на уроджений гіпотиреоз підвищена частота алелей HLA: Bw44, Aw24, B18. У 5–10 % причиною вродженого гіпотиреозу є дисгормоногенез (генетичні дефекти синтезу, секреції або периферичного метаболізму тиреоїдних гормонів). Мутація гена бета-субодиниці ТТГ описана як рідкісна причина його дефіциту.
Отже, варто відзначити, що на сьогодні причини і механізми розвитку низки ендокринних захворювань залишаються не до кінця вивченими, і очікуване широке впровадження в практику нових технологій молекулярно-генетичного аналізу, включаючи високопродуктивні системи паралельного секвенування, безумовно, дозволить добитися подальшого прогресування в генетичній ендокринології.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Тюльпаков А.Н. Роль молекулярной генетики в диагностике и лечении эндокринных заболеваний. Краткая характеристика моногенных форм наследственных эндокринопатий, диагностированных в ФГУ ЭНЦ за 15-летний период (1996-2010) / А.Н. Тюльпаков // Проблемы эндокринологии. — 2011. — № 1. — С. 28-35.
2. Giordano M. Genetic causes of isolated and combined pituitary hormone deficiency / М. Giordano // Best Pract. Res. Clin. Endocrinol. Metab. — 2016. — Vol. 30(6). — Р. 679-691. doi: 1016/j.beem.2016.09.005.
3. Rostomyan L. Screening for genetic causes of growth hormone hypersecretion / L. Rostomyan, A. Beckers // Growth Horm IGF Res. — 2016. — Vol. 30–31. — Р. 52-57. doi: 10.1016/j.ghir.2016.10.004.
4. Aslan I.R. The melanocortin system and insulin resistance in humans: insights from a patient with complete POMC deficiency and type 1 diabetes mellitus / I.R. Aslan, S.A. Ranadive,
I. Valle [et al.] // J. Obes. (Lond.). — 2014. — Vol. 38(1). — Р. 148-51. doi: 10.1038/ijo.2013.53.
5. Тюльпаков А.Н. Клиническая и молекулярно-генетическая характеристика 10 случаев врожденной гипоплазии надпочечников, обусловленной дефектами фактора DAX1 / А.Н. Тюльпаков, Н.Ю. Калинченко // Проблемы эндокринологии. — 2010. — № 2. — P. 3-9.
6. Дзеранова Л.К. Новая мутация в гене CYP17: описание клинического случая / Л.К. Дзеранова, А.Н. Тюльпаков, Е.А. Пигарова [и др.] // Проблемы эндокринологии. — 2006. — № 6. — С. 41-44.
7. Амирасланова М.М. Роль дефицита 21-гидроксилазы в генезе врожденной дисфункции коры надпочечников / М.М. Амирасланова, Н.Ф. Мамедова, Е.А. Соснова // Научные ведомости Белгородского государственного университета. Серия: Медицина. Фармация. — 2011. — Т. 14. — Вып. № 10(105). — С. 44-47.
8. Зелинская Н.Б. Состояние гормонального статуса детей с врожденной дисфункцией коры надпочечников, обу–словленной дефицитом 21-гидроксилазы / Н.Б. Зелинская, Н.Л. Погадаева, Е.В. Глоба, И.Ю. Шевченко, Т.Н. Бегутова, О.А. Хорошая, В.Б. Малашонок // Перинатология и педиатрия. — 2015. — № 1. — С. 54-59.
9. Shozu M. Understanding the pathological manifestations of aromatase excess syndrome: lessons for clinical diagnosis / M. Shozu, M. Fukami, T. Ogata // Expert. Rev. Endocrinol. Metab. — 2014. — Vol. 9(4). — Р. 397-409. doi: 10.1586/
17446651.2014.926810.
10. Калинченко Н.Ю. Клиническое описание и молекулярно-генетическая верификация 2 случаев изолированной минералокортикоидной недостаточности вследствие дефицита альдостеронсинтазы / Н.Ю. Калинченко, Н.А. Зубкова, А.Н. Тюльпаков // Проблемы эндокринологии. — 2009. — № 1. — С. 28-31.
11. Foster S.R. G protein-coupled receptors in cardiac biology: old and new receptors / S.R. Foster, E. Roura, P. Molenaar, W.G. Thomas // Biophys. Rev. — 2015. — Vol. 7(1). — Р. 77-89. doi: 1007/s12551-014-0154-2.
12. Michigami T. Disorders Caused by Mutations in Calcium-Sensing Receptor and Related Diseases / Т. Michigami // Clin. Calcium. — 2017. — Vol. 27(4). — Р. 521-527.
doi: CliCa1704521527.
13. Duan K. Clinicopathological correlates of hyperparathyroidism / K. Duan, K. Gomez Hernandez, O. Mete // J. Clin. Pathol. — 2015. — Vol. 68(10). — Р. 771-87. doi: 10.1136/jclinpath-2015-203186.
14. Витебская А.В. Случай тяжелого неонатального гиперпаратиреоза, обусловленный дефектом кальций-сенсорного рецептора / А.В. Витебская, Е.Е. Петряйкина, А.Ю. Разумовский [и др.] // Проблемы эндокринологии. — 2010. — № 4. — С. 27-33.
15. Hannan F.M. Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites / F.M. Hannan, M.A. Nesbit, C. Zhang [et al.] // Hum. Molec. Genet. — 2012. — Vol. 21. — Р. 2768-2778 [PubMed: 22422767].
16. Inden M. The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains / М. Inden, M. Iriyama, M. Zennami // Brain Res. — 2016. — Vol. 15(1637). — Р. 128-36. doi: 10.1016/j.brainres.2016.02.032.
17. Ларина А.А. Аутоиммунные полигландулярные синдромы взроcлых: генетические и иммунологические критерии диагностики / А.А. Ларина, Е.А. Трошина, О.Н. Иванова // Проблемы эндокринологии. — 2014. — № 60(3). — С. 43-52. doi: 10.14341/probl201460343-52.
18. Turton J.P. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency / J.P. Turton, R. Reynaud, A. Mehta [et al.] // J. Clin. Endocrinol. Metab. — 2005. — Vol. 90(8). — Р. 4762-4770. doi: https://doi.org/10.1210/jc.2005-0570.
19. Колодкина А.А. Клиническая, гормональная и молекулярно-генетическая характеристики трех случаев нарушения формирования пола (НФП) 46XY, обусловленного дефицитом 5-редуктазы II типа / А.А. Колодкина, М.Е. Карманов, Н.Ю. Калинченко [и др.] // Проблемы эндокринологии. — 2010. — № 3. — С. 34-40.
20. Bonomi M. New understandings of the genetic basis of isolated idiopathic central hypogonadism / M. Bonomi, D.V. Libri, F. Guizzardiet [et al.] // Asian Journal of Andrology. — 2012. — Vol. 14(1). — Р. 49-56. doi: 10.1038/aja.2011.68.
21. Кураева Т.Л. Генетика моногенных форм сахарного диабета / Т.Л. Кураева, Л.И. Зильберман, Е.В. Титович, В.А. Петеркова // Сахарный диабет. — 2011. — № 1. — С. 20-27.
22. Зелінська Н.Б. Статистика цукрового діабету в дітей в Україні (аналіз та прогноз) / Н.Б. Зелінська, Є.В. Глоба, Н.Л. Погадаєва // Клінічна ендокринологія та ендокринна хірургія. — 2013. — № 1(42). — С. 80-83.
23. Дедов И.И. MODY тип 2: клинические и молекулярно-генетические характеристики 13 случаев заболевания. Первое описание MODY в России / И.И. Дедов, Н.А. Зубкова, Н.Ю. Арбатская [и др.] // Проблемы эндокринологии. — 2009. — № 3. — С. 3-7.
24. Ellard S. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing / S. Ellard, H. Lango Allen, E. De Franco [et al.] // Diabetologia. — 2013. — Vol. 56. — Р. 1958-1963. doi: 10.1007/s00125-013-2962-5.
25. Global IDF/ISPAD Guideline for diabetes in childhood and adolescence // Pediatr Diabetes. — 2009. — Vol. 10(Suppl. 12). — Р. 1-210.
26. Глоба Є.В. Неонатальний цукровий діабет в Украї–ні: сучасні діагностичні та терапевтичні можливості / Є.В. Глоба // Клінічна ендокринологія та ендокринна хірургія. — 2015. — № 2(50). — С. 71-75.
27. Alsaedi K.A. Extended clinical features associated with novel Glis3 mutation: a case report / K.A. Alsaedi, A.B. Alghamdi, A. Aljasser, A. Altawil, M. Naglaa Kamal Alghamdi [et al.] // BMC Endocrine Disorders. — 2017. — Vol. 17. — Р. 14. doi: 10.1186/s12902-017-0160-z.
28. Farooqi S. Genetics of obesity in humans / S. Farooqi, S. O’Rahilly // Endocr. Rev. — 2006. — Vol. 27(7). — P. 710-18. doi: 10.1210/er.2006-0040.
29. Heianza Y. Gene-Diet Interaction and Precision Nutrition in Obesity / Y. Heianza, L. Qi // Int. J. Mol. Sci. — 2017. — Vol. 7(18). — Р. 787. doi: 10.3390/ijms18040787.
30. Петеркова В.А. Неиммунный тиреотоксикоз, обусловленный активирующей мутацией гена рецептора тиреотропного гормона (первое описание в России) / В.А Петеркова, О.В. Васюкова, А.Н. Тюльпаков // Проблемы эндокринологии. — 2009. — № 2. — С. 48-50.
31. Hannoush Z.C. Defects of Thyroid Hormone Synthesis and Action / Z.C. Hannoush, R.E. Weiss // Еndocrinol. Metab. Clin. North Am. — 2017. — Vol. 46(2). — Р. 375-388. doi: 10.1016/j.ecl.2017.01.005.
32. Wang F. Next-generation sequencing of NKX2.1, FOXE1, PAX8, NKX2.5, and TSHR in 100 Chinese patients with congenital hypothyroidism and athyreosis / F. Wang, C. Liu, X. Jia [et al.] // Clin. Chim. Acta. — 2017. — Vol. 25(470). — Р. 36-41. doi: 10.1016/j.cca.2017.04.020.