В практической деятельности врача-эндокринолога нередко встречаются ситуации, когда у больного наблюдаются поражения двух и более эндокринных желез с характерными проявлениями их гипо- или гиперфункции. Часто первичная хроническая надпочечниковая недостаточность (ХНН) сочетается с другими аутоиммунными эндокринопатиями, что получило название аутоиммунного полигландулярного синдрома (АПС).
Впервые взаимосвязь между недостаточностью надпочечниковых желез и тиреоидитом обнаружил Н. Schmidt в 1926 году, и это сочетание болезней впоследствии получило в литературе название «синдром Шмидта». Значительно позже, в 1964 году, С. Carpenter и соавт. сообщили о сочетании первичной ХНН и аутоиммунного тиреоидита (АИТ) с инсулинозависимым сахарным диабетом (СД), и этот вариант сочетания аутоиммунных эндокринопатий носит название «синдром Карпентерa».
Термин «АПС» был впервые введен в 1981 году М. Neufeld и соавт. с выделением кардинальных различий АПС 1-го и 2-го типов. АПС 2-го типа определен как сочетание первичной ХНН с АИТ и/или СД 1-го типа при отсутствии гипопаратиреоза и кожно-слизистого кандидоза. АПС 2-го типа с точки зрения формальной генетики является аутосомно-доминантным заболеванием с неполной пенетрантностью, однако возможны спорадические случаи возникновения заболевания.
Синдром возникает наиболее часто в возрасте от 20 до 30 лет, у женщин в 8 раз чаще, чем у мужчин. Клиническое течение отдельных болезней не отличается от таковых в случае возникновения самостоятельной нозологической формы. Особый интерес с клинической точки зрения представляют наблюдения больных с синдромом полиэндокринопатии в случае сочетания первичной ХНН и СД, а также первичной ХНН, АИТ и СД 1-го типа. Сочетание СД и первичной ХНН встречается не очень часто. Однако такие случаи имеются, при этом СД и первичная ХНН могут возникать одновременно или СД присоединяется к ранее развившейся первичной ХНН. В большинстве случаев СД 1-го типа развивается раньше, чем первичная ХНН.
Van Buchen (1950) описал 27 случаев сочетания СД и первичной ХНН, из них у 16 СД развился до манифестации надпочечниковой недостаточности, у 6 — после и у 5 — одновременно.
В ряде работ показано, что высокие уровни –аутоантител обнаруживаются в субклинической стадии первичной ХНН с последующим снижением их в период выраженных клинических проявлений. Поэтому, возможно, больные СД 1-го типа с высоким уровнем аутоантител к 21-гидроксилазе могут иметь субклиническую фазу первичной ХНН. Длительные наблюдения за этими больными позволили установить, что субклиническая фаза первичной ХНН может продолжаться несколько лет, при этом клиническая манифестация заболевания развивается в среднем через 5 лет после появления антител к ткани коры надпочечников. Интервал между клиническим проявлением его отдельных компонентов может составить более 20 лет. У 40–50 % больных с исходно изолированной первичной ХНН рано или поздно развивается другая аутоиммунная эндокринопатия. В противоположность этому у лиц, страдающих аутоиммунной патологией щитовидной железы при отсутствии в семейном анамнезе АПС 2-го типа, риск развития сочетанной эндокринной патологии относительно низок.
В клинической картине у больных АПС 2-го типа превалируют проявления первичной ХНН, гиперпигментация при этом может быть выражена слабо, особенно при сочетании первичной ХНН и гипотиреоза. Если СД присоединяется к ранее существовавшей первичной ХНН, то свидетельством проявления заболевания служат полиурия и полидипсия. У таких больных очень высокая чувствительность к инсулину. В случае присоединения первичной ХНН к ранее существовавшему СД 1-го типа довольно часто наблюдается картина мнимого улучшения обмена веществ и снижение дозы инсулина. В литературе описаны подобные случаи. Так, S. Simpson (1949) описал случай развития первичной ХНН у больного 27 лет, который страдал инсулинозависимым СД с 11 лет. За 5 лет до смерти развилась первичная ХНН. Доза инсулина снизилась сначала с 72 до 48 ЕД/сут, а затем до 6 ЕД/сут. При этом отмечались резкие колебания гликемии — 45–1540 мг% (2,5–75 ммоль/л).
D. Bernstein описал случай сочетания СД и первичной ХНН с типичными клиническими проявлениями с последующим присоединением гипотиреоза (синдром Карпентера), что представляло значительные трудности для дифференциальной диагностики с гипопитуитаризмом.
Приводим случай собственного наблюдения одного из вариантов течения АПС 2-го типа — синдрома Карпентера, который имел некоторые клинические особенности течения и представлял трудности для диагностики и лечения.
Больная Т., 25 лет, поступила в эндокринологическое отделение областной больницы (история болезни № 12 227) с диагнозом «сахарный диабет 1-го типа, тяжелая форма, лабильное течение со склонностью к гипогликемиям, в состоянии декомпенсации. Диабетическая ангиопатия нижних конечностей. Аутоиммунный тиреоидит. Гипотиреоз».
При поступлении больная предъявляла жалобы на потерю аппетита, тошноту, периодическую рвоту, которая не приносила облегчения, сильную головную боль, ухудшение памяти, выраженную слабость, частые гипогликемические состояния, потерю массы тела (минус 6 кг за последние 2 месяца), снижение артериального давления (АД) до 90–60 мм рт.ст., парестезии и боль в ногах, потемнение кожи с характером выраженного равномерного загара.
Анамнез заболевания. Со слов пациентки, болеет СД 1-го типа около 3,5 года. В октябре 2001 г. в возрасте 21 года в связи с угрозой прерывания беременности (27-я неделя) находилась в гинекологическом отделении ЦРБ на лечении. Больную беспокоили кровотечения мажущего характера, причиной которых было предлежание плаценты. При исследовании содержания сахара в крови обнаружено его увеличение до 7,5–8 ммоль/л. Однако на это повышение гликемии лечащие врачи не обратили внимания, расценив его как «физиологические изменения, характерные для беременности». Больная не была проконсультирована врачом-эндокринологом и никакого лечения не проводилось, несмотря на то, что больную беспокоила умеренная жажда. Через месяц после родов жажда усилилась, появилась сухость во рту, беспокоил обильный диурез. К врачу не обращалась, состояние продолжало ухудшаться, что сопровождалось потерей массы тела. Через 4 месяца после родов было проведено повторное исследование содержания сахара в крови и обнаружено его увеличение до 18 ммоль/л, а также глюкозурия. Определение ацетона в моче не проводилось. Поставлен диагноз «СД 1-го типа» и назначено лечение инсулином в дозе 60 ЕД/сут (актрапид НМ 20 ЕД и протафан НМ 40 ЕД). Несколько месяцев спустя в связи с недостаточной эффективностью лечения и плохим самочувствием была направлена в эндокринологическое отделение областной клинической больницы, где была проведена коррекция дозы инсулина и подтвержден диагноз СД 1-го типа. Кроме этого, в связи с наличием в анамнезе указаний на заболевание щитовидной железы было проведено определение уровня тиреоидных гормонов (трийодтиронина (Т3), тироксина) и тиреотропного гормона (ТТГ). Со слов больной, уровень ТТГ равнялся 15 мЕД/л, установлен диагноз послеродового гипотиреоза. Назначено лечение левотироксином в дозе 50 мкг/сут. Спустя некоторое время (3–4 месяца) было проведено определение уровня ТТГ, показатели которого составляли 0,15 мЕД/л. После заочной консультации эндокринолога прекратила прием левотироксина. Самочувствие было удовлетворительным до декабря 2004 года, когда значительно участились гипогликемические состояния, появилась слабость, кровоточивость из десен, отмечалась пигментация десен, языка. Летом после отдыха в Куяльнике у больной отмечался сильный равномерный загар, который сохранялся всю зиму, в то время как у родственников он исчез. По поводу заболевания десен лечилась у стоматолога, однако без заметного успеха. В феврале 2005 года в связи с тяжелыми гипогликемическими состояниями 3–4 раза в сутки и резкими колебаниями уровня сахара в крови по собственной инициативе обратилась в диабетологический отдел Института эндокринологии и обмена веществ имени В.П. Комиссаренко НАМН Украины. В процессе стационарного обследования было обнаружено увеличение содержания ТТГ до 29,7 мЕД/л и уменьшение содержания свободного Т3, а также снижение экскреции с мочой 11-оксикортикостероидов (ОКС) — 0,13 мкмоль/л (норма от 0,18 до 0,8 мкмоль/л), 17-ОКС — 18,1 мкмоль/л (норма от 17,0 до 62 мкмоль/л). Повторное исследование экскреции 11-ОКС и 17-ОКС с мочой подтвердило низкие показатели. Больной выставлен диагноз АИТ и манифестного гипотиреоза и назначено лечение левотироксином в дозе 100 мкг/сут. В отделении больная проконсультирована стоматологом-пародонтологом, который указал на возможность заболевания надпочечников, однако это не было принято к сведению лечащим врачом.
14.03.2005 больная выписана из отделения, однако состояние было средней тяжести. Во время поездки в поезде появилась рвота. Дома состояние продолжалось ухудшаться, появились вышеуказанные жалобы. Госпитализирована в терапевтическое отделение ЦРБ. Лечащим врачом на основании –изучения выписки из предыдущего лечебного учреждения и снижения экскреции с мочой количества 11-ОКС и 17-ОКС под вопросом диагностирована надпочечниковая недостаточность. Для дальнейшего лечения и уточнения диагноза больная была направлена в эндокринологическое отделение областной клинической больницы.
Анамнез жизни. Росла и развивалась в детстве нормально. Материальные условия жизни были удовле–творительными. В школе училась хорошо. Перенесла ветряную оспу. В детстве страдала упорным фурункулезом. В возрасте 12 лет при очередном профилактическом осмотре обнаружено увеличение щитовидной железы, по поводу чего обследовалась в детской областной клинической больнице. Диагноз и лечение не помнит. Мать страдает гипертонической болезнью. Сахарным диабетом страдала прабабушка по материнской линии, а с отцовской стороны родная тетя страдала выраженным ожирением и сахарным диабетом 2-го типа. Месячные с 12 лет, регулярные, редкие. Беременности — 2, роды — 1, аборт — 1.
Аллергию к пище и медикаментам отрицает. Не курит, алкоголем не злоупотребляет, наркотические средства не принимает. Инвалид второй группы по общему заболеванию.
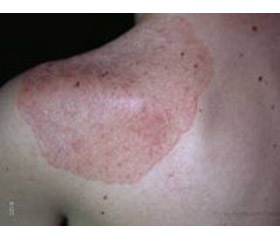
Объективное состояние на момент госпитализации. Состояние средней тяжести. Рост — 167 см, масса тела — 62 кг. Правильного телосложения, несколько пониженного питания. Мышечная система развита слабо. Тургор кожи снижен, кожа сухая, равномерно пигментирована на открытых и закрытых участках тела. Отмечается выраженная пигментация губ, десен, боковых поверхностей языка. Значительно пигментированы наружные гениталии и перианальная область. Пульс — 84 в 1 мин, ритмичный, слабого наполнения и напряжения, АД — 90/60 мм рт.ст. Границы сердца в пределах нормы. Тоны сердца приглушены, деятельность ритмична. Перкуторно над легкими ясный легочной звук, аускультативно в легких везикулярное дыхание. Живот мягкий, болезненный в эпигастральной области. Печень и селезенка не пальпируются. Щитовидная железа видна при осмотре при нормальном положении шеи. Пальпаторно отмечается диффузное увеличение железы, она безболезненна, подвижна при глотании, плотно-эластичной консистенции. Со стороны других органов и систем физикально изменений не обнаружено.
Предварительный диагноз: сахарный диабет 1-го типа, тяжелая форма, состояние декомпенсации. Лабильное течение со склонностью к гипогликемиям и кетоацидозу. Аутоиммунный тиреоидит, гипертрофическая форма с исходом в манифестный гипоти–реоз. Хроническая надпочечниковая недостаточность впервые выявленная, в состоянии декомпенсации.
Результаты проведенного лабораторно-инструментального обследования. Общий анализ крови: Нb — 137 г/л, Эр. — 4,6 × 1012/л, Л. — 5,0 × 109/л, п. — 4, с. — 46, л. — 32, э. — 8, м. — 2, скорость оседания эритроцитов — 8 мм/ч. Общий анализ мочи: сахар — 1 %, в остальном — без особенностей. Био–химические исследования: общий белок — 75 г/л, мочевина — 5,7 ммоль/л, креатинин — 75 мкмоль/л, общий билирубин — 16,3 мкмоль/л, аспартатаминотрансфераза — 0,2, аланинаминотрансфераза — 0,4 млмоль/л/ч, тимоловая проба — 3,1 ЕД, калий сыворотки — 3,5 ммоль/л, натрий — 137 ммоль/л. Гормональные исследования: ТТГ — 17 мЕД/л, кортизол крови — 65 ммоль/л (норма от 150 до 660 ммоль/л). Глюкоза крови натощак: 23,0–9,6–22,5–21,2–23,6–22,0–10,6–9,9 ммоль/л. Гликемический профиль — от 3,9 до 20,0 ммоль/л. Глюкоза мочи — 1 %, ацетон мочи — не выявлен. Ультразвуковое исследование (УЗИ) щитовидной железы от 22.03.2004: объем правой доли — 15,9 см3, левой — 13,2 см3. Объем долей представлен рыхлой неоднородной тканью с участками различной плотности неправильной формы и фиброзной тяжистостью без четкого узлообразования. Картина аутоиммунного тиреоидита?
Приводим данные УЗИ щитовидной железы трехгодичной давности (20.01.2001) с целью оценки динамики: объем правой доли — 32,8 см3, левой — 17,0 см3. Щитовидная железа увеличена больше за счет правой доли, контуры ровные, структура диффузно уплотнена.
Эхоэнцефалография без патологии. Реоэнцефалография: кровенаполнение снижено в районе вертебробазилярного бассейна. Сосудистый тонус повышен. Венозный отток вялый, с выраженным застоем во всех бассейнах. Венозная дисфункция.
Консультации специалистов. Окулист: умеренная инъекция слизистой. Глазное дно: диск зрительного нерва сохранен, бледный, артерии и вены узкие. Диагноз «макулярный отек сетчатки».
Невропатолог: дисметаболическая энцефалопатия второй степени. Стойкий цефалгический синдром. Ликвородинамические кризы.
Клинический диагноз. На основании жалоб больной, анамнеза болезни и жизни, объективного исследования, данных лабораторно-инструментальных методов обследования установлен диагноз «аутоиммунный полигландулярный синдром 2-го типа (вариант течения синдром Карпентера) — сахарный диабет 1-го типа, тяжелая форма, лабильное течение со склонностью к гипогликемии и кетоацидозу, состояние декомпенсации. Аутоиммунный тиреоидит, гипертрофическая форма с исходом в манифестный гипотиреоз. Первичная хроническая надпочечниковая недостаточность, классический вариант течения, тяжелая форма, состояние декомпенсации».
Лечение. Проводилась заместительная терапия гидрокортизоном по 50 мг 3 раза в день внутримышечно, внутривенные инфузии раствора Рингера, витаминотерапия. Затем осуществлен постепенный переход на таблетированные препараты кортизона ацетата 25 мг утром и 12,5 мг вечером, кортинефф — по 100 мкг утром. Наряду с этим проводилась заместительная терапия левотироксином в дозе 100 мкг/сут и интенсифицированная терапия инсулином (базальный инсулин протафан НМ 12 ЕД в 8:30, прандиальный инсулин новорапид: 12 ЕД — в 8:30, 8 ЕД — в 14:00, 12 ЕД — в 18:00. Общая доза — 60 Ед/сут).
Доза инсулина после назначения глюкокортикоидов увеличилась с 44 до 60 ЕД/сут. По мере увеличения дозы глюкокортикоидов наблюдались резкие колебания гипергликемии и рост дозы инсулина. На фоне проведенного лечения у больной увеличилась масса тела на 3 кг, значительно уменьшилась пигментация кожи и слизистых, отмечался прилив сил.
Для приведенного случая синдрома Карпентера характерны некоторые особенности течения. Они заключаются в следующем:
1. Дебют заболевания (АПС 2-го типа) начался в подростковом возрасте с АИТ, который проявился увеличением щитовидной железы.
2. Выраженная пигментация кожи и слизистых на фоне исходного манифестного гипотиреоза. Принято считать, что в данном случае она выражена слабо.
3. Практически одновременное (с учетом ла–тент–ного периода первичной ХНН) развитие сахарного диабета 1-го типа и надпочечниковой недостаточности.
4. В качестве фактора, способствующего манифестации ассоциированных с синдромом заболеваний, выступило такое состояние, как беременность.
Врачу-эндокринологу следует помнить о том, что ухудшение контроля гликемии (лабильное течение СД) может свидетельствовать о присоединении первичной ХНН при наличии других признаков заболевания. Особое внимание следует уделять склонности к гипогликемии и высокой чувствительности к инсулину.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.