
Международный эндокринологический журнал Том 14, №7, 2018
Вернуться к номеру
Роль експресії гена Foxo1 у механізмі антигіпертрофічної дії метформіну в кардіоміоцитах
Авторы: Пасєчко Н.В., Лой Г.Я., Корда М.М., Олещук О.М.
ДВНЗ «Тернопільський державний медичний університет ім. І.Я. Горбачевського МОЗ України», м. Тернопіль, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Актуальність. Діабетична кардіоміопатія є провідною причиною смертності при цукровому діабеті (ЦД) 2-го типу. Однією з основних патоморфологічних ознак розвитку діабетичної кардіоміопатії розглядається гіпертрофія кардіоміоцитів. Метформін, засіб вибору першої лінії для лікування ЦД 2-го типу, окрім антигіперглікемізуючих, викликає кардіопротективні ефекти. Проте механізм дії препарату в кардіоміоцитах вивчений недостатньо. Мета. Вивчення ролі експресії гена Foxo1 у механізмі антигіпертрофічної дії метформіну в кардіоміоцитах. Матеріали та методи. Н9С2 клітини були трансфектовані за допомогою siRNA Foxo1 та siRNA negative control. Клітини були депривовані у 0% медіумі протягом 24 годин, за 30 хвилин до стресу проліковані метформіном (5 mM) і поміщені у гіпоксичний бокс на 16 годин із подальшою реоксигенацією протягом чотирьох годин. Площі клітин визначені за допомогою програми ImageJ. Ефективність трансфекції підтверджено за допомогою полімеразної ланцюгової реакції в реальному часі. Результати. При нормальному функціонуванні гена Fохо1 метформін проявляє виражену антигіпертрофічну дію в умовах гіпоксії. Проте блокування експресії Fохо1 позбавляє препарат цього ефекту і викликає гіпертрофію Н9С2 клітин у всіх умовах експерименту. Висновки. Виражена гіпертрофічна відповідь у групі клітин, трансфектованих за допомогою siRNA Foxo1, які культивувалися в умовах гіпоксії і були проліковані метформіном, може бути результатом одного з механізмів: а) метформін запобігає збільшенню розмірів клітин через Fохо1-залежний механізм, тому блокування гена Fохо1 призводить до втрати препаратом антигіпертрофічного ефекту; б) метформін захищає від гіпоксії незалежно від Fохо1-шляху, тому виражена гіпертрофія лікованих метформіном клітин, інкубованих у гіпоксії, є наслідком гальмування експресії Fохо1, що є потужним гіпертрофічним стимулом. Отже, з’ясування кардіопротективного механізму дії метформіну залишається надзвичайно актуальним питанням і потребує подальшого вивчення.
Актуальность. Диабетическая кардиомиопатия (ДКМ) является ведущей причиной смертности при сахарном диабете (СД) 2-го типа. Одним из основных патоморфологических признаков развития ДКМ считается гипертрофия кардиомиоцитов. Метформин, препарат выбора первой линии для лечения СД 2-го типа, кроме гипогликемических, вызывает кардиопротекторные эффекты. Однако механизм действия препарата в кардиомиоцитах изучен недостаточно. Цель. Изучение роли экспрессии гена Foxo1 в механизме антигипертрофического действия препарата метформин в кардиомиоцитах. Материалы и методы. Н9С2 клетки были трансфектированы с помощью siRNA Foxo1 и siRNA negative control. Клетки были депривированы в 0% медиуме в течение 24 часов, за 30 минут до стресса пролечены метформином (5 mM) и помещены в гипоксический бокс на 16 часов с последующей реоксигенацией в течение четырех часов. Площади клеток определяли с помощью программы ImageJ. Эффективность трансфекции подтверждено с помощью полимеразной цепной реакции в реальном времени. Результаты. При нормальном функционировании гена Fохо1 метформин оказывает выраженное антигипертрофическое действие в условиях гипоксии. Однако блокировка экспрессии гена Fохо1 лишает препарат этого эффекта и вызывает гипертрофию Н9С2 клеток во всех условиях эксперимента. Выводы. Выраженная гипертрофическая реакция в группе клеток, трансфектированих с помощью siRNA Foxo1, которые культивировались в условиях гипоксии и были пролечены метформином, может быть результатом одного из механизмов: а) метформин предупреждает увеличение размеров клеток через Fохо1-зависимый механизм, поэтому блокировка гена Fохо1 приводит к потере препаратом антигипертрофического эффекта; б) метформин защищает от гипоксии независимо от Fохо1-пути, поэтому выраженная гипертрофия леченных метформином клеток, инкубированных в гипоксии, является следствием торможения экспрессии Fохо1, что является мощным гипертрофическим стимулом. Итак, изучение кардиопротекторного механизма действия метформина остается чрезвычайно актуальным вопросом и требует дальнейшего изучения.
Background. Diabetic cardiomyopathy is the leading cause of mortality in patients with type 2 diabetes mellitus. Hypertrophy of cardiomyocytes is one of the main pathomorphological signs of diabetic cardiomyopathy development. Metformin, the first-line drug for the treatment of type 2 diabetes mellitus, along with hypoglycaemic effects, exerts cardioprotective effects. However, the mechanism of metformin action in cardiomyocytes remains unclear. The purpose of the study was to investigate the role of Foxo1 gene expression in the mechanism of antihypertrophic action of metformin in cardiomyocytes. Materials and methods. H9C2 cells were transfected with siRNA Fохо1 and siRNA negative control. Cells were deprived in 0% medium for 24 hours, treated with metformin (5mM) 30 min before cell stress, then put into hypoxic chamber for 16 hours and reoxygenated for 4 hours. Cell area was quantified using ImageJ. Knockdown efficiency was confirmed by real time polymerase chain reaction. Results. At the normal functioning of Fохо1 gene, metformin has the expressed antihypertrophic action under hypoxia. However, blocking Fохо1 gene expression deprives preparation of this effect and causes the hypertrophy of Н9С2 cells in all conditions of the experiment. Conclusions. The strong hypertrophic response in the group of H9C2 cells transfected with siRNA Foxo1 cultured under hypoxia with metformin treatment may be a result of following mechanisms: a) metformin prevents hypertrophy through Foxo1 pathway, thus, Foxo1 silencing totally blocked metformin protective effects on H9C2 hypertrophy; b) metformin protects against hypoxia independently of Foxo1 pathway, therefore, strong hypertrophy of metformin-treated cells incubated in hypoxia is the result of Foxo1 knockdown, a potent hypertrophic stimulus. Consequently, further investigations are still required to clarify the mechanisms by which metformin exerts its cardioprotective effects.
діабетична кардіоміопатія; гіпертрофія; Н9С2 клітини; метформін; Fохо1
диабетическая кардиомиопатия; гипертрофия; Н9С2 клетки; метформин; Fохо1
diabetic cardiomyopathy; hypertrophy; H9C2 cells; metformin; Fохо1
Вступ
Всесвітня організація охорони здоров’я (ВООЗ) визначає цукровий діабет (ЦД) як одне з чотирьох пріоритетних неінфекційних захворювань, оскільки число його випадків і поширеність невпинно зростають. У 2014 році від ЦД страждали 422 мільйони дорослого населення планети [1]. На ЦД 2–го типу припадає 90–95 % усіх випадків захворювання [2].
ЦД призводить до формування мікро– та макросудинних уражень, негативно впливає практично на всі тканини в організмі, викликає виражену дисфункцію внутрішніх органів із подальшою інвалідизацією хворих, яка, у свою чергу, стає соціальним та економічним тягарем у всьому світі. У структурі смертності від серцево–судинних захворювань (ССЗ) понад 65 % становлять випадки, пов’язані з ЦД [3].
Діабетичне ураження серця найчастіше проявляється у вигляді ішемічної хвороби серця (ІХС), серцевої вегетативної нейропатії та діабетичної кардіоміопатії (ДКМ) [3]. Саме діабетична кардіоміопатія є однією з основних причин термінальної стадії серцевої недостатності (СН), що часто призводить до раптової зупинки серця та смерті [4]. А з–поміж усіх інших коморбідних із СН станів найвищою була смертність при ДКМ, яка спостерігалася у 60 % пацієнтів [5]. Фактична поширеність ДКМ ще не встановлена через відсутність достатньої кількості даних про дослідження серед різних груп населення, які страждають від ЦД [3].
Патогенез і патофізіологія ДКМ не вивчені достатньо. Відомо, що для цього виду кардіоміопатії характерною є фокальна втрата клітин внаслідок мікросудинного спазму та реперфузного ушкодження з подальшим розвитком фокального фіброзу та реактивної гіпертрофії у відповідь на некроз міокарда [6]. Гістологічно ДКМ характеризується апоптозом, гіпертрофією та фіброзом, функціонально–кардіальною дисфункцією, що виникають незалежно від судинних або серцевих захворювань [7].
Вважається, що хронічна гіперглікемія відіграє центральну роль у розвитку патології. Разом із тим численні комплексні (на початку компенсаторні, а потім патологічні) механізми, взаємодія багатьох молекулярних і метаболічних процесів у міокарді та плазмі крові сприяють патогенезу ураження [3]. Саме гіперглікемія спричиняє значні метаболічні та молекулярні зміни у кардіоміоцитах. Підвищений метаболізм глюкози призводить до посилення оксидативного стресу шляхом утворення активних форм кисню у мітохондріях [8]. Надмірна продукція супероксиду мітохондріальним дихальним ланцюгом викликає зниження скоротливої здатності міокарда, що зрештою призводить до фіброзу кардіоміоцитів [9], ушкодження ДНК і прискореного апоптозу кардіоміоцитів [3]. У свою чергу, гіперінсулінемія є важливим тригером розвитку гіпертрофії серця при ДКМ [10].
Гіпертрофія міокарда є однією з найбільш поширених ознак ДКМ [6]. Характерною рисою гіпертрофованих кардіоміоцитів є збільшення їх розмірів порівняно з нормальними клітинами. Дослідження культивованих in vitro неонатальних кардіоміоцитів показали, що збільшення розмірів кардіоміоцитів під впливом гіпертрофічних стимулів пов’язане з посиленням саркомерогенезу та збільшенням експресії натрійуретичних пептидів [9].
Як і в попередні роки, у новому алгоритмі терапії ЦД 2–го типу Американської діабетичної асоціації зразка 2018 року пероральний цукрознижуючий засіб метформін із групи бігуанідів рекомендований як препарат першої лінії для стартової фармакотерапії ЦД 2–го типу у дітей і дорослих за відсутності протипоказань. Крім того, метформін залишається препаратом вибору з точки зору ефективності та безпеки для монотерапії ЦД 2–го типу [10]. Найбільшими його клінічними перевагами є те, що він не викликає гіпоглікемій і збільшення маси тіла. Метформін характеризується високою антигіперглікемічною ефективністю і має високий профіль серцево–судинної безпеки [11].
Попри те, що препарат у клініці почали застосовувати ще в 1950–х роках, точний механізм дії метформіну досі залишається недостатньо вивченим [12]. Насамперед метформін підвищує чутливість рецепторів до інсуліну, що призводить до зниження його рівня у плазмі крові натще та зменшення інсулінорезистентності [13], підвищує рівень глюкагоноподібного пептиду–1 (GLP–1) у плазмі крові та індукує експресію гена інкретинового рецептора острівця Лангерганса через залежний від пероксисомних проліфератор––активованих рецепторів (PPAR)–α механізм [14].
Проте основна роль відводиться здатності метформіну знижувати продукцію глюкози печінкою шляхом пригнічення глюконеогенезу [15]. Вивчаються також інші можливі інгібіторні механізми на печінковий глюконеогенез, зокрема зміни ферментної активності або зменшення захвату печінкою глюконеогенних субстратів.
Упродовж багатьох років молекулярна мішень метформіну залишалася невідомою [12], поки група вчених [16] не дослідила, що активація AMP–активованої протеїнкінази (AMPK) була тісно пов’язана з плейотропними ефектами метформіну.
AMPK є багатокомпонентним ферментом, а саме основним регулятором ліпідних шляхів біосинтезу, завдяки своїй ролі у фосфорилюванні й інактивації ключових ензимів, таких як ацетил–КоА–карбоксилаза. AMPK–активована протеїнкіназа вважається сенсором енергетичного обміну, оскільки вона є чутливою до клітинного співвідношення АМФ : АТФ [17]. Будучи активованою, AMPK переводить клітини з анаболічного до катаболічного стану, виключаючи синтетичні шляхи, що споживають АТФ, і відновлює енергетичний баланс. Як основний внутрішньоклітинний датчик енергії AMPK стає важливою мішенню при метаболічних порушеннях, особливо при ЦД 2–го типу та захворюваннях печінки. Через це гіпоглікемізуючий ефект метформіну пов’язаний з активацією печінкової АМРК [18].
У дослідженнях останніх років вивчаються нові альтернативні шляхи, що, ймовірно, залучені до контролю метформіном гомеостазу глюкози [19]. Наприклад, метформін пригнічує активність AMP–деамінази (AMPD). Виключення експресії AMPD нівелювало стимуляцію метформіном транспортування глюкози [20].
Хоча більшість досліджень показують, що метформін проявляє свої кардіопротективні властивості шляхом активації АМФ–активованої протеїнкінази [21], доведено, що наявні інші потенційні механізми дії препарату, незалежні від цього ферменту [22].
Fохо–група протеїнів включає Fохо1, Fохо3a, Fохо4 та Fохо6, які забезпечують різноманітні фізіологічні та патологічні функції, регулюючи експресію генів, пов’язаних із апоптозом, зупинкою клітинного циклу, репарацією ушкодженої ДНК, метаболізмом глюкози, стійкістю до оксидативного стресу та іншими функціями [23].
Отже, ДКМ, основними чинниками розвитку якої є гіперглікемія та гіпоксія, посідає перше місце серед причин смертності у пацієнтів із ЦД 2–го типу. Тому вивчення цього діабетичного ускладнення та його патоморфологічних ознак, зокрема гіпертрофії, апоптозу, некрозу, фіброзу та запалення, залишається надзвичайно актуальним. Необхідно детальніше вивчити шляхи, завдяки яким метформін проявляє свою захисну дію на серцеві клітини.
Мета дослідження: дослідити, чи залучена експресія гена Foxo1 до механізму антигіпертрофічної дії метформіну в кардіоміоцитах.
Матеріали та методи
Експеримент був проведений на Н9С2 культурі клітин (кардіоміобласти ембріонів щурів). Н9С2 клітини були культивовані у середовищі Dulbecco’s Modified Eagle’s medium (DMEM, Life Technologies) із додаванням 10% сироватки (Fetal Bovine Serum, Life Technologies) та пеніцилін–стрептоміцинової суміші (Penicillin–Streptomycin 10,000 U/mL, Life Technologies) в СО2–інкубаторі при 37 °С в атмосфері 5% CO2 і вологості повітря 95 %. Коли конфлюентність клітин сягла 40–60 %, було проведено трансфекцію клітин за допомогою Fохо1 small interfering RNA (siRNA) та siRNA negative control (NC) у кінцевій концентрації 200 nmol/L з використанням Lipofectamine 2000 transfection reagent (Life Technologies) відповідно до інструкції виробника. Через 48 години подальшої інкубації конфлюентність клітин сягала 80–90 % і вони були готові до подальших експериментів.
Щоб здійснити депривацію клітин, живильне середовище було замінене на Dulbecco’s Modified Eagle’s medium із пеніцилін–стрептоміциновою сумішшю, але без додавання сироватки (0% medium). Через 24 години, за 30 хвилин до стресу (гіпоксії), клітини були проліковані метформіном у кінцевій концентрації 5 mM.
Щоб викликати гіпоксію, клітини були поміщені у гіпоксичний бокс (5% CO2, 1% O2, balance N2) при 37 °C на 16 годин із подальшою реоксигенацією протягом чотирьох годин.
Щоб визначити площу клітин, випадковим чином було обрано по 3 поля зору для кожної умови і використано програму ImageJ. Ефективність трансфекції було підтверджено за допомогою полімеразної ланцюгової реакції в реальному часі.
Дані наведені у вигляді mean ± SEM. Статистична обробка здійснена за допомогою one–way ANOVA followed by a Bonferroni’s post hoc test у програмі GraphPad Prism version 5.00 (GraphPad Software, Inc).
Результати
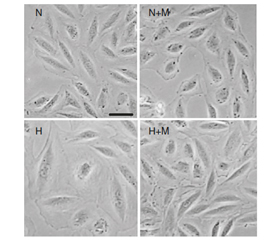
На рис. 1 продемонстровано, що культивування кардіоміоцитів ембріонів щура в умовах гіпоксії (Н) вірогідно викликає збільшення їх площі як у випадку фізіологічної експресії гена Fохо1 (при застосуванні siRNA Сontrol), так і при його блокуванні за допомогою siRNA Fохо1.
/707-1.jpg)
Лікування метформіном вірогідно захищає Н9С2 клітини від індукованої гіпоксією гіпертрофії при нормальному функціонуванні гена Fохо1 (Н + М + siRNA Сontrol).
Проте у випадку блокування гена Fохо1 (при трансфекції Н9С2 клітин за допомогою siRNA Fохо1) препарат метформін втрачав свою властивість запобігати збільшенню площі кардіоміоцитів в умовах гіпоксії (Н + М + siRNA Fохо1).
Зважаючи на ці результати, можна було б припустити, що Fохо1–шлях є потенційним механізмом, через який метформін реалізує свій антигіпертрофічний ефект у Н9С2 клітинах, оскільки виключення експресії гена Fохо1 призводить до втрати препаратом здатності запобігати збільшенню площі кардіоміоцитів під впливом гіпоксії.
Проте блокування гена Fохо1 призвело до гіпертрофії Н9С2 клітин і в усіх інших умовах, навіть при нормоксії (N + siRNA Fохо1 та N + М + siRNA Fохо1). Це свідчить про те, що виключення Fохо1–експресії провокує збільшення площі клітин, тобто нормальне функціонування гена є обов’язковою умовою для підтримки фізіологічних розмірів кардіоміоцитів.
Як показано на рис. 2, рівень експресії гена Fохо1 після трансфекції Н9С2 клітин із siRNA Fохо1 вірогідно зменшився. Незначну експресію гена Fохо1 у групах трансфектованих із siRNA Fохо1 клітин можна пояснити тим, що до етапу трансфекції клітини були у конфлюентності 40–60 % і експресували ген Fохо1.
/708-1.jpg)
Обговорення
Культури кардіоміоцитів, отримані зазвичай від новонароджених щурів, широко використовуються для дослідження клітинних і молекулярних змін, що лежать в основі гіпертрофічної відповіді. Культивування кардіоміоцитів дозволило створити доступну, з хорошими характеристиками in vitro систему для вивчення гіпертрофічної відповіді. Клітинна лінія H9C2 отримана з ембріональної тканини шлуночків, що є надзвичайно важливим фактором, оскільки серцева гіпертрофія, спричинена артеріальною гіпертензією, переважно виникає у міокарді лівого шлуночка [24].
Слід відзначити, що H9C2 клітини здатні проявляти основні гіпертрофічні ознаки, спричинені гіпертрофічними стимулами in vitro [25]. У дослідженні групи вчених [24] було підтверджено, що неонатальні кардіоміоцити щурів імітують широкий спектр гіпертрофічних ознак і мають великі перспективи для аналізу функції кардіоміоцитів in vitro.
Вважається, що кардіальна гіпертрофія є адаптивним механізмом. У відповідь на механічні, гемодинамічні, гормональні та патологічні подразники серце пристосовується до підвищених потреб для серцевої діяльності шляхом збільшення м’язової маси через ініціювання гіпертрофічної реакції. Хоча гіпертрофічна відповідь спочатку є компенсаторним механізмом, стійка гіпертрофія може призвести до дилатаційної кардіоміопатії, СН і раптової смерті. Кінцевим наслідком кардіальної гіпертрофії є СН із дилатацією шлуночка та прогресуючим зниженням серцевого викиду, що призводить до вентрикулярної дисфункції та злоякісної аритмії. Гіпертрофія — фінальна молекулярна реакція кардіоміоцитів на різноманітні гіпертрофічні сигнали. Отже, кардіальна гіпертрофія є важливим фактором ризику СН [26].
На молекулярному рівні гіпертрофія кардіоміоцитів вважається результатом дисбалансу між прогіпертрофічними й антигіпертрофічними факторами та їх механізмами, що контролюють ріст клітин [26].
На клітинному рівні гіпертрофія кардіоміоцитів характеризується збільшенням розмірів клітин, посиленням синтезу білків і підвищеною організацією саркомеру [9].
Механізм виникнення кардіальної гіпертрофії при ЦД 2–го типу полягає у тому, що на тлі системної гіперінсулінемії, що є результатом ІР, посилюються ефекти інсуліну в клітинах, які не втрачають чутливості до гормону, зокрема у кардіоміоцитах. Отже, гіпертрофія є проявом мітогенної дії інсуліну [27].
Здатність метформіну пригнічувати синтез протеїнів у кардіоміоцитах, зменшувати серцеву масу та експресію прогіпертрофічних цитокінів свідчить про його терапевтичну ефективність при кардіальній гіпертрофії. Здатність метформіну зменшувати ізопротеренол–індуковану гіпертрофію кардіоміо–цитів було підтверджено морфометрично. Крім того, метформін зменшував підвищену ізопротеренолом експресію профібротичних генів. Доведено, що метформін ефективно зменшує гіпертрофію, викликану підвищеним тиском [28].
У своєму дослідженні Evans et al. продемонстрували, що у пацієнтів із ЦД 2–го типу та СН, які приймали метформін або його комбінацію з препаратами сульфонілсечовини, спостерігався значно нижчий рівень смертності, порівняно з тими, хто отримував монотерапію препаратами сульфонілсечовини [29]. Інша група вчених (Aguilar et al.) підтвердила, що виживання пацієнтів, які отримували терапію метформіном, було істотно вищим [30]. Romero et al. [31] продемонстрували, що у пацієнтів із ЦД та СН лікування метформіном асоціюється з нижчими рівнями смертності та госпіталізації.
Дослідження UKPDS показало, що лікування метформіном, попри схожі з іншими цукрознижуючими засобами показники глікемічного контролю, асоціювалося зі зменшенням рівня загальної смертності та смерті від ЦД, а також із нижчим ризиком інфаркту міокарда [11]. У дослідженні SPREAD–DIMCAD встановлено, що пацієнти із ЦД 2–го типу та кардіоваскулярними захворюваннями, ліковані метформіном, мали на 46 % менший показник серцево–судинних ускладнень порівняно з тими, хто отримував гліпізид [32].
Наявні дані, що метформін може втручатися у ренін–ангіотензин–альдостеронову систему, індукуючи АМРК, внаслідок чого пригнічується синтез ангіотензину ІІ [33]. При індукованій ангіотензином ІІ гіпертрофії культивованих кардіоміоцитів метформін проявляв антигіпертрофічні властивості [34].
Відомо, що метформін чинить свої ефекти шляхом активації AMPK, яка інтенсивно експресується в печінці, кишечнику, скелетних м’язах, а також у міокарді [35].
Група дослідників Hu M. et al. [36] у своєму in vitro дослідженні виявила, що метформін у низькій дозі (5 mM) захищає H9C2 клітини (кардіоміоцити ембріонів щурів) від ушкодження, викликаного гіпоксією та гіперглікемією. Кардіопротективний ефект препарату in vitro був реалізований через AMPK/JNK–залежний механізм.
Проте доведено, що метформін чинить кардіопротективні ефекти також незалежно від AMPK [22].
В літературі описано, що метформін діє в ендотеліальних клітинах [37], печінці [38], ракових клітинах ендометрію шляхом регуляції активності транс–крипційного білка forkhead box O1 (Fохо1), який кодується однойменним геном [39]. Встановлено також, що Fохо1 відіграє неабияку роль у підтримці кардіального гомеостазу [40].
У процесі експерименту нами було підтверджено, що метформін має виражені кардіопротективні властивості, зокрема, ефективно запобігає гіпер–трофії Н9С2 клітин. Наше дослідження також доводить, що експресія гена Fохо1 є критичним фактором у підтримці фізіологічних розмірів кардіо–міоцитів.
Разом із тим на сьогодні нез’ясованим залишається питання, чи залучена експресія гена Fохо1 у механізмі антигіпертрофічної дії метформіну.
Висновки
В результаті проведених нами досліджень встановлено, що при нормальному функціонуванні гена Fохо1 метформін проявляє виражену антигіпертрофічну дію в умовах гіпоксії. Проте блокування експресії Fохо1 позбавляє препарат цього ефекту і викликає гіпертрофію Н9С2 клітин в усіх умовах експерименту.
На нашу думку, виражена гіпертрофічна відповідь у групі клітин (Н + М + siRNA Fохо1) може бути результатом одного з механізмів:
— метформін запобігає збільшенню розмірів клітин через Fохо1–залежний механізм, тому блокування гена Fохо1 призводить до втрати препаратом антигіпертрофічного ефекту;
— метформін захищає від гіпоксії незалежно від Fохо1–шляху, тому виражена гіпертрофія лікованих метформіном клітин, інкубованих у гіпоксії, є наслідком гальмування експресії Fохо1, що є потужним гіпертрофічним стимулом.
Отже, з’ясування кардіопротективного механізму дії метформіну є надзвичайно актуальним питання і залишається остаточно не вивченим.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Джерело фінансування — програма Erasmus+.
Подяки. Дослідження проведене в лабораторії Інституту кардіоваскулярних і метаболічних захворювань (Institut des Maladies Métaboliques et Cardiovasculaires (I2MC — UMR1048)) в рамках угоди про співпрацю між ДВНЗ «Тернопільський державний медичний університет ім. І.Я. Горбачевського МОЗ України» та Paul Sabatier University, Toulouse, Франція. Висловлюємо подяку за допомогу у проведенні дослідження команді № 14 під керівництвом Dr. Oxana Kunduzova.
Інформація про внесок кожного автора: виконання досліджень, статистична обробка результатів — Лой Г.Я.; аналіз результатів — Лой Г.Я., Олещук О.М., Корда М.М.; актуальність — Пасєчко Н.В.