
Международный эндокринологический журнал Том 15, №3, 2019
Вернуться к номеру
Синдром Клайнфельтера у дітей і підлітків: поєднання генетики й ендокринології
Авторы: Сорокман Т.В., Попелюк Н.О., Макарова О.В.
ВДНЗ України «Буковинський державний медичний університет», м. Чернівці, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Метою роботи було навести огляд даних літератури щодо етіології і патогенезу синдрому Клайнфельтера (СК). Нами проведений огляд наукової літератури щодо СК за ключовими словами «синдром Клайнфельтера — Klinefelter syndrome», «синдром Клайнфельтера — Рейфенштейна — Олбрайта — Klinefelter-Reifenstein-Albright syndrome», «пубертатне недорозвинення сім’явивідних канальців — pubertal underdevelopment of seminiferous tubules», «синдром 47, XXY — syndrome 47, XXY» з використанням PubMed як пошукової системи. Статистичні дані щодо СК неоднозначні. Окремими авторами частота народження осіб із СК визначена як 1 випадок на 500–1000 живонароджених хлопчиків, надалі зростає до 3–4 % серед безплідних чоловіків, у пацієнтів із азооспермією діагностується у 10–12 % випадків. Приблизно 10 % випадків СК виявляються шляхом пренатальної діагностики. Фенотипова мінливість може залежати від ступеня вираженості генетичних дефектів, дефіциту андрогенів, чутливості андрогенних рецепторів (CAG-поліморфізм) або від випадкової інактивації додаткового матеріалу X-хромосоми. Педіатри повинні знати про фенотипову мінливість синдрому Клайнфельтера, зокрема звертати увагу на психологічні та вербальні порушення. Необхідно також проводити комплексне молекулярно-генетичне дослідження з медико-генетичним консультуванням усіх дітей із неоднозначними статевими органами. Вчасно призначена терапія зможе мінімізувати не тільки комплекс фенотипових проявів синдрому, але й, можливо, відстрочити швидку втрату клітин сперматогенезу, що дозволить допомогти цим пацієнтам у реалізації їх репродуктивної функції.
Целью работы было представить обзор данных литературы по этиологии и патогенезу синдрома Клайнфельтера (СК). Нами проведен обзор научной литературы по ключевым словам «синдром Клайнфельтера — Klinefelter syndrome», «синдром Клайнфельтера — Рейфенштейна — Олбрайта — Klinefelter-Reifenstein-Albright syndrome», «пубертатное недоразвитие семявыносящих канальцев — pubertal underdevelopment of seminiferous tubules», «синдром 47, XXY — syndrome 47, XXY» с использованием PubMed в качестве поисковой системы. Статистические данные по СК неоднозначны. Отдельными авторами частота рождения лиц с СК определена как 1 случай на 500–1000 живорожденных мальчиков, продолжает расти до 3–4 % среди бесплодных мужчин, у пациентов с азооспермией диагностируется в 10–12 % случаев. Примерно 10 % случаев СК диагностируются пренатально. Фенотипическая изменчивость может зависеть от степени выраженности генетических дефектов, дефицита андрогенов, чувствительности андрогенных рецепторов (CAG-полиморфизм) или от случайной инактивации дополнительного материала X-хромосомы. Педиатры должны знать о фенотипической изменчивости синдрома Клайнфельтера, в частности обращать внимание на психологические и вербальные нарушения. Необходимо также проводить комплексное молекулярно-генетическое исследование с медико-генетическим консультированием всех детей с неоднозначными половыми органами. Вовремя назначенная терапия сможет минимизировать не только комплекс фенотипических проявлений синдрома, но и, возможно, отсрочить быструю потерю клеток сперматогенеза, что позволит помочь этим пациентам в реализации их репродуктивной функции.
The purpose was to analyze literature on the etiology and pathogenesis of Klinefelter syndrome (KS). The review of scientific literature regarding KS by the key words “Klinefelter syndrome”, Klinefelter-Reifenstein-Albright syndrome”, “pubertal underdevelopment of seminiferous tubules”, “syndrome 47, XXY” using PubMed as a search engine. Statistical data on KS are ambiguous. According to some authors, the frequency of births of children with KS defined as 1 case per 500–1000 live-birth boys further increases to 3–4 % among infertile men, and in patients with azoospermia, KS is diagnosed in 10–12 % of cases. Approximately 10 % of cases of KS is detected by prenatal diagnosis. Phenotypic variability may depend on the severity of genetic defects, androgen deficiency, androgen receptor sensitivity (CAG polymorphism) or accidental inactivation of the additional information of the X chromosome. Pediatricians should be aware of the phenotypic variability of the KS, in particular, to pay attention to mental and verbal disorders. It is also necessary to conduct a comprehensive molecular genetic study with medical genetic counseling for all children with ambiguous sexual organs. Timely prescribed therapy will able to minimize not only phenotypic manifestations of the syndrome, but also possibly postpone the rapid loss of spermatogenic cells, which will help these patients to realize their reproductive function.
синдром Клайнфельтера; клінічний і генетичний поліморфізм; огляд
синдром Клайнфельтера; клинический и генетический полиморфизм; обзор
Klinefelter syndrome; clinical and genetic polymorphism; review
Вступ
Останніми роками кількість дітей, народжених із різними захворюваннями, діагностика яких при народженні утруднена, поступово зростає [1, 2]. Особливо це стосується тих патологічних станів, які зазвичай діагностуються цитогенетичним методом, оскільки в нашій країні каріотипування в періоді новонародженості проводиться тільки в окремих випадках. У зв’язку з викладеним вище вивчення захворювань, пов’язаних із утрудненою діагностикою в ранньому віці, є надзвичайно актуальним. До таких захворювань належить синдром Клайнфельтера (СК), який посідає третє місце серед усього спектра ендокринних захворювань. Випереджають його лише цукровий діабет і патологія щитоподібної залози [3].
Метою роботи було навести огляд даних літератури щодо етіології і патогенезу СК.
Огляд
Нами проведений огляд наукової літератури щодо СК за ключовими словами «синдром Клайнфельтера — Klinefelter syndrome», «синдром Клайнфельтера — Рейфенштейна — Олбрайта — Klinefelter-Reifenstein-Albright syndrome», «пубертатне недорозвинення сім’явивідних канальців — pubertal underdevelopment of seminiferous tubules», «синдром 47, XXY — syndrome 47, XXY» з використанням PubMed як пошукової системи. Беручи до уваги дослідження, проведені за останні 10 років, проаналізовані реферати 275 статей. Критерій для відбору статей для дослідження ґрунтувався на їх тісній актуальності з темою. Більш детально вивчено результати дослідження, висвітлені у 45 статтях.
Синоніми синдрому: синдром Клайнфельтера — Рейфенштейна — Олбрайта, синдром Клайнфельтера, пубертатне недорозвинення сім’явивідних канальців (Міжнародна класифікація хвороб 10-го перегляду: Q98.0–Q98.2, Q98.4; MedlinePlus: 000382; eMedicine: ped/1252 MeSH: D007713).
СК — генетичне захворювання, що характеризується наявністю додаткової жіночої статевої хромосоми Х (однієї або декількох) у чоловічому каріотипі ХY і виявляється в першу чергу ендокринними порушеннями за типом первинного чоловічого гіпогонадизму (недостатності утворення статевих гормонів безпосередньо в чоловічих статевих залозах), частіше виявляється в період статевого дозрівання.
Учені розходяться в думці, коли і ким синдром уперше був описаний [4, 5]. Так, є дані про те, що вперше синдром описано Altmann в 1895 р. і Berblinger в 1934 р. Однак клінічне і патологоанатомічне вивчення синдрому як самостійної нозологічної форми та спільна публікація в 1942 р. вперше належать Г.Ф. Клайнфельтеру молодшому (Harry Fitch Klinefelter Jr) з колегами (Рейфенштейн Е.С. — Reifenstein Е.С. та Олбрайт Ф. — Albright Fuller) у звіті щодо обстеження 9 чоловіків, об’єднаних спільними симптомами, такими як гінекомастія, азоспермія без алейдигізму, підвищена екскреція фолікулостимулюючого гормону [6, 7]. Цитогенетична основа СК вперше була описана Р. Бріггсом (Robert Briggs) і М. Баром (Murray Llewellyn Barr) в 1956 році. У каріотипі хворих вони виявили зайву Х-хромосому — каріотип був 47, XXY. Інші джерела вказують на приналежність опису цитогенетичної основи СК в 1959 році П.А. Джейкобс (Patricia Ann Jacobs) та Дж.А. Стронгу (John Anderson Strong), які працювали в клініці Western General Hospital в Единбурзі, Шотландія. Цей каріотип (47, ХХY) належав 24-річному чоловікові, у якого були ознаки СК [8]. Надалі з’явилися повідомлення і про інші типи полісомії, об’єднані під загальною назвою — синдром Клайнфельтера. Так, Ferguson-Smith et al. (1960) і Carr et al. (1961) повідомили про випадок трисомії за Х-хромосомою — 48, ХХХY; Muldal і Ockey (1960), Carr et al. (1961) — за 48, ХХYY; Ford et al. (1959), Crooke і Hayward (1960) першими опублікували дані щодо мозаїчних форм СК — 47, ХХY/46, Х; Fraccaro et al. (1960), Anders et al. (1960), Miller et al. (1961) повідомили про випадок тетрасомії за Х-хромосомою — 49, ХХХХYY [9].
Додаткова хромосома X залишається у зв’язку з нерозходженням хромосом під час мейозу I (гаметогенезу). Нерозходження відбувається тоді, коли гомологічні хромосоми, в даному випадку статеві X- і Y-хромосоми, не розділяються, утворюючи сперматозоїди з Х- і Y-хромосомами відповідно. Внаслідок чого звичайно нормальна (Х) запліднена яйцеклітина призводить до народження XXY-потомства. Інший механізм збереження додаткової хромосоми X — жіночий і пов’язаний із нерозходженням хромосом під час мейозу II. Нерозходження відбуватиметься тоді, коли сестринські хроматиди статевих хромосом, у цьому випадку XX, не розділяються. У такому випадку яйцеклітина буде мати набір ХХ-хромосом, і після запліднення сперматозоїдом Y з’явиться XXY-нащадок. Отже, помилки в прогенезі найчастіше виникають при нерозходженні хромосом в мейозі I, а в разі материнського походження — ще й у мейозі II (рис. 1, 2).
/224-1.jpg)
Статистичні дані щодо СК неоднозначні. Окремими авторами частота народження осіб із СК визначена як 1 випадок на 500–1000 живонароджених хлопчиків, надалі зростає до 3–4 % серед безплідних чоловіків, у пацієнтів із азооспермією діагностується у 10–12 % випадків [10]. Відзначається, що частота захворюваності у чоловіків із нормальним фенотипом становить 1 : 1100 чоловіків, серед хворих на шизофренію — 1 : 300, олігофренію — 1 : 95 випадків. Оскільки СК практично в 100 % випадків супроводжується безплідністю, за статистикою, в одного із 10 чоловіків, які страждають від безплідності, виявляється СК [11, 12]. Однак тільки 10 % випадків СК діагностуються в дитячому віці.
Точні причини виникнення захворювання на сьогодні не встановлені. Більшість дослідників заперечують фактор впливу спадковості, при цьому відзначають фактори ризику, що можуть сприяти розвитку патології: занадто молодий або, навпаки, дуже зрілий вік матері, несприятлива екологічна обстановка, шлюб між кровними родичами.
Пренатальна діагностика містить неінвазивні скринінгові методи: пренатальний ДНК-тест Pano–rama (Illumina, США) і протеомний тест. При виконанні тесту Panorama з крові матері (починаючи з 9-го тижня вагітності) виокремлюється і досліджується ДНК плода, що дозволяє отримати вірогідні результати порівняно зі звичайним біохімічним скринінгом. При протеомному тесті досліджується кров жінки в II триместрі вагітності на вміст церулоплазміну, аполіпопротеїну і плазмаретинолзв’язуючого білка [13]. Скринінгові методи визначають тільки ймовірність розвитку СК, діагностика ж СК як у пренатальному (біопсія ворсин хоріона, кордоцентез, амніоцентез), так і постнатальному (лімфоцити крові) періодах проводиться цитогенетичним аналізом. Приблизно 10 % випадків СК виявляється шляхом пренатальної діагностики [14].
Типова клінічна картина 47, XXY-синдрому (високорослість, євнухоїдна статура, порушення формування вторинних статевих ознак, гіпоплазія яєчок, азооспермія, гінекомастія, рання еректильна дисфункція) розвивається в період статевого дозрівання і в постпубертатному віці, однак запідозрити синдром якомога раніше з певною часткою ймовірності при підозрі на мозаїчний варіант СК можливо лише у деяких дітей із помірною розумовою недостатністю за наявності в лініях проаналізованих клітин додаткової статевої хромосоми. До початку статевого розвитку вдається відзначити тільки окремі фізичні ознаки: довгі ноги, висока талія, високий зріст, але на відміну від типового євнухоїдизму розмах рук у них рідко перевищує довжину тіла, ноги помітно довші тулуба. Пік надбавки зросту припадає на період між 5–8 роками, і середній зріст дорослих пацієнтів становить приблизно 179,2 ± 6,2 см. Окремі автори вказують, що у пацієнтів із СК відзначається знижений обсяг яєчок до періоду статевого дозрівання. У підлітковому віці синдром найчастіше проявляється збільшенням грудних залоз, хоча в деяких випадках цієї ознаки може й не бути. При цьому необхідно зазначити, що у 60–75 % підлітків пубертатного віку також відзначається збільшення грудних залоз — пубертатна гінекомастія, яка, однак, самостійно проходить протягом двох років, у той час як у пацієнтів із СК гінекомастія зберігається на все життя, вона двостороння і, як правило, безболісна.
У деяких пацієнтів із СК знижений інтелект і обмежені вербальні і пізнавальні здібності. Коефіцієнт інтелекту (IQ) у таких пацієнтів перебуває в широких межах. Однак вербальний коефіцієнт зазвичай нижче пізнавального. Легка розумова відсталість при СК трапляється у 25–50 % випадків, але іноді буває і більш тяжкий ступінь [15, 16]. Через знижену продукцію андрогенів часто розвиваються остеопороз і м’язова слабкість. Нерідко спостерігаються ожиріння, порушення толерантності до глюкози і цукровий діабет 2-го типу. У чоловіків із СК частота автоімунних захворювань значно вище порівняно із здоровими. Є повідомлення про підвищену частоту розвитку ревматоїдного артриту, системного червоного вовчака та інших системних колагенозів, автоімунних захворювань щитоподібної залози [17–23].
При мозаїцизмі (46, XY/47, XXY) клінічні симптоми виражені слабо, і окремі хворі можуть зберігати, хоча і знижену, здатність до запліднення [24].
Пацієнти із СК (48, XXYY) відрізняються більш високим зростом, а решта клінічних проявів нічим не відрізняються від пацієнтів з каріотипом 47, XXY. Зазвичай такі пацієнти характеризуються як тихі і скромні, проте можуть бути агресивними й імпульсивними, у них відзначаються набагато нижчі адаптивні можливості в соціальному середовищі. В осіб із СК (48, XXXY) часто відзначаються такі аномалії, як очний гіпертелоризм, плоске перенісся, променево-ліктьовий синостоз, клинодактилія п’ятого пальця. Коефіцієнт інтелекту зазвичай знаходиться в межах 40–60, мовлення таких хворих значно уповільнене. У поведінці відзначається виражений інфантилізм.
Більш виражені порушення фізичного і розумового розвитку мають особи із СК (49, XXXXY). Вони проявляються мікроцефалією, очним гіпертелоризмом, плоским переніссям, вузькими очними щілинами. Зріст таких хворих зазвичай низький. Вони можуть також мати щілину піднебіння, вади серця (в тому числі відкритий ductus arteriosus), променево-ліктьовий синостоз, вальгусне викривлення колінних суглобів, деформацію стоп, клинодактилію п’ятого пальця. Обсяг яєчок, а також розмір статевого члена у таких пацієнтів маленькі. IQ знижений і знаходиться в межах 20–60. Їх зазвичай описують як скромних і доброзичливих, з випадковими нападами дратівливості і спалахами гніву, мають труднощі в адаптації до постійно змінюваних умов соціального середовища [25–30].
Для з’ясування причини відмінностей фенотипового прояву синдрому проведено молекулярний аналіз першого екзона гена андрогенового рецептора [31, 32]. Зазначений ген складається з восьми екзонів, перший із яких кодує трансактивний домен, другий і третій — ДНК-зв’язуючий домен, 5’-кінець четвертого екзона — залежний домен, 3’-кінець четвертого екзона спільно з п’ятим і шостим екзонами кодує ліганд-зв’язуючий домен. При цьому саме перший екзон містить поліморфні повтори CAG, що кодують поліглутамінові ланцюги андрогенового рецептора. Встановлено взаємозв’язок між різницею числа повторів і експресією андрогенового CAG-рецептора (у середньому 20 повторів). З огляду на те, що ген андрогенового рецептора знаходиться на Х-хромосомі, яка в нормі у чоловіків є в єдиному екземплярі, а у жінок одна з них інактивована, природно, що відсутність лайонізації призводить до додаткової експресії генів зазначеної хромосоми, пов’язаних із структурними функціями головного мозку. Крім того, присутність Pars-генів Y-хромосоми, які мають аналоги або не мають аналогів у Х-хромосомі, в результаті подвоєння Х-хромосоми у хворих із СК збільшує дозу експресії, що також впливає на фенотипову варіабельність клінічних проявів захворювання [33].
Розширений аналіз експресії генів Х-хромосоми показав, що майже 15 % генів уникають інактивації [34–36]. Відомо, що гени, які уникають інактивації, розташовані не довільним чином, а на короткому плечі Х-хромосоми, і 10 % із них мають змінну інактивацію, тобто в одних жінок вони заблоковані, а в інших — ні. Аналогічна ситуація спостерігається й у чоловіків із СК: надлишкова Х-хромосома інактивується, але не тотально. Ті гени, які уникли інактивації, мають вищий рівень експресії у чоловіків із СК порівняно з чоловіками з каріотипом 46, XY. При цьому підвищені дози генних продуктів у пацієнтів із СК теж можуть бути різними, що призводить до широкого спектра клінічних і фенотипових проявів захворювання. Так, при повногеномному SNP-генотипуванні (Single nucleotide polymorphism), коли можна знайти відмінності послідовності ДНК розміром в один нуклеотид між гомологічними ділянками гомологічних хромосом, установлено варіації числа копій генів (CNVs) у більш ніж 40 % пацієнтів із СК, що виникають внаслідок незбалансованих хромосомних перебудов. Виявлено, що CNVs у пацієнтів із СК у 94,4 % випадків виявляються дуплікаціями генів. До цих генів відносяться SHOX, CSF2RA, SLC25A6, PCDH11X і IL9R, які розміщені в псевдоавтосомальних ділянках (PARs) і Xq21.31. Вищевказані гени мають гомологи на Y-хромосомі, що означає присутність двох копій у здорових чоловіків і жінок, трьох копій — у пацієнтів із СК без дуплікації і чотирьох копій — із дуплікацією. Гени STS та PNPLA4 присутні тільки на Х-хромосомі [37]. Ці дані узгоджуються з результатами іншого дослідження [38]. На даний час встановлено не тільки гени, що піддаються дуплікації у пацієнтів із СК, але і генні продукти, а також метаболічні шляхи, де вони задіяні. Окрім збільшення генних продуктів, Х-хромосома впливає і на експресію генів автосом. Аналіз ДНК лімфоцитів у пацієнтів із СК з використанням ДНК мікрочіпів показав, що 480 генів автосом мали підвищену експресію, а близько 200 — знижену [39].
Таким чином, фенотип хворого з синдромом Клайнфельтера залежить від числа зайвих Х-хромосом і довжини CAG-алелі [40–42]. Підвищення цих показників прямо пропорційне психічному розвитку, соціальній адаптації, професійній діяльності і зворотно пропорційне розвитку чоловічих статевих ознак.
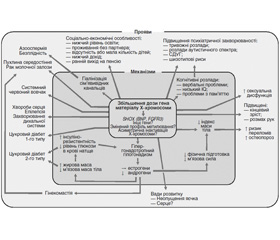
Вплив підвищеної дози генів на Х-хромосому (і, можливо, інші генетичні механізми, такі як інша картина метилювання і асиметрична X-інактивація, що працює узгоджено) перебуває в центрі сучасного розуміння патогенезу СК (рис. 3). Це призводить до низки ефектів, що впливають на ендокринну систему, особливо гіпергонадотропного гіпогонадизму, зміни складу тіла і підвищеного ризику остеопорозу. Гіпогонадизм має поширені ефекти, що впливають: 1) на різні рівні гормонів; 2) серцево-судинні ознаки; 3) метаболічні особливості; 4) безплідність. Гіпогонадизм призводить до хибного кола, що підвищує резистентність до інсуліну і посилює несприятливі зміни складу тіла, прямо чи опосередковано. Зростає поінформованість щодо поганого соціально-економічного профілю та психіатричної захворюваності.
/226-1.jpg)
При збільшенні числа Х-хромосом ряд генів експресується в гермінативних клітинах яєчка, що може впливати на мейотичний розподіл і відігравати роль в етіології безплідності чоловіків із СК [44]. Це підтверджується фактом збільшення експресії специфічного білка TEX11 (білка гермінативних клітин яєчка, кодованого генами Х-хромосоми), який найбільш чітко виявляється в сперматогоніях і ранніх сперматоцитах та проявляється пригніченням клітинної проліферації в сім’яних канальцях [45].
Ускладнення, що можуть стати наслідком СК, включають проблеми з розумовим розвитком, які можуть розвинутися до стадії дебільності; психічні порушення, що призводять до алкоголізму, суїцидальних настроїв, асоціального способу життя; високий ризик розвитку цукрового діабету, ожиріння; уроджених вад серця; остеопорозу (крихкість кісток); ймовірність виникнення злоякісних утворень (табл. 1) [43].
/227-1.jpg)
Висновки
Синдром Клайнфельтера залишається знач–ною мірою недіагностованим у дитячому віці. Фенотипова мінливість може залежати від ступеня вираженості генетичних дефектів, дефіциту андрогенів, чутливості андрогенних рецепторів (CAG-поліморфізм) або від випадкової інактивації додаткового матеріалу X-хромосоми. Педіатри повинні знати про фенотипову мінливість синдрому Клайнфельтера, зокрема, звертати увагу на психологічні та вербальні порушення. Необхідно також проводити комплексне молекулярно-генетичне дослідження з медико-генетичним консультуванням усіх дітей із неоднозначними статевими органами. Вчасно призначена терапія зможе мінімізувати не тільки комплекс фенотипових проявів синдрому, але й, можливо, відстрочити швидку втрату клітин сперматогенезу, що дозволить допомогти цим пацієнтам у реалізації їх репродуктивної функції.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.