
Международный эндокринологический журнал Том 17, №4, 2021
Вернуться к номеру
Адреногенітальний синдром у чоловіків: класична форма. Клінічний випадок
Авторы: Ляшук П.М.(1), Станкова Н.І.(2), Ляшук Р.П.(1)
(1) — Буковинський державний медичний університет, м. Чернівці, Україна
(2) — Чернівецький обласний ендокринологічний центр, м. Чернівці, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
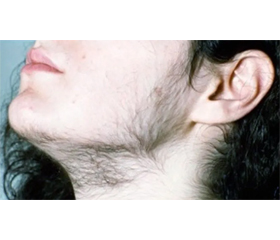
Останнім часом у зарубіжній науковій літературі появилися повідомлення, що в хлопчиків і юнаків із класичною вірильною формою адреногенітального синдрому (АГС) або вродженою дисфункцією кори надниркових залоз у результаті неадекватної замісної глюкокортикоїдної терапії у 21–28 % випадків трапляється об’ємне утворення в яєчках із залишкової надниркової тканини (Testicular Adrenal Rest Tumors — ТАRТ), що збільшується під впливом надмірної продукції адренокортикотропного гормона (АКТГ). Дане доброякісне утворення розміром до 2 см і більше в діаметрі виявляється пальпаторно і при проведенні ультразвукового дослідження (УЗД). Утворення можуть тиснути на тканину яєчок і призвести до гіпогонадизму. Такі особи можуть мати низький рівень тестостерону через зниження функції клітин Лейдіга. ТАRТ зазвичай зменшується після оптимізації лікування. Іноді у зв’язку з підозрою на онкопроцес проводять необґрунтоване оперативне втручання. Наведено клінічний випадок класичної форми уродженого АГС, що проявився передчасним ізосексуальним статевим дозріванням, крипторхізмом та рідкісними доброякісними об’ємними утвореннями в яєчках із залишків надниркової тканини, що збільшилися під впливом надмірної продукції АКТГ у результаті неадекватної замісної терапії глюкокортикоїдними препаратами. Утворення виявляються при проведенні УЗД, зменшуються при оптимізації лікування. Спостереження за пацієнтом в динаміці показало, що одними з основних діагностичних гормональних тестів є рівні в крові АКТГ і 17-гідроксипрогестерону, які на момент виявлення захворювання були надмірно високими. Постійна замісна терапія глюкокортикоїдними препаратами підтримує рівень даних показників у межах референтних значень. Своєчасна діагностика характеру патології, постійна коригуюча гормональна терапія забезпечили здатність пацієнта адаптуватися в житті та суспільстві відповідно до свого статусу. Клінічних проявів гіпокортицизму та/або гіперандрогенії в батьків нашого пацієнта не виявлено, що свідчить про автосомно-рецесивне успадкування АГС. У перспективі важливим є генетичне консультування майбутніх батьків, особливо з проявами гіперандрогенії, для оцінки можливого розвитку подібної патології в їх потомства.
Recently, in the foreign scientific literature there have been reports that boys and young men with the classic virile form of congenital adrenal hyperplasia or congenital dysfunction of the adrenal cortex as a result of inadequate glucocorticoid therapy in 21–28 % of cases have testicular adrenal rest tumors, which increases under the influence of excessive production of adrenocorticotropic hormone (ACTH). This benign formation up to 2 cm in diameter and larger is detected by palpation and ultrasound. The formations can press on the testicular tissue and lead to hypogonadism. Such individuals may have low testosterone levels due to decreased Leydig cell function. Testicular adrenal rest tumors usually decrease after treatment optimization. Unreasonable surgery is sometimes performed in suspected cancer. A clinical case is presented of the classic form of congenital adrenal hyperplasia, manifested itself in isosexual precocious puberty, cryptorchidism and testicular adrenal rest tumors rare, increased under the influence of excessive ACTH production as a result of inadequate glucocorticoid replacement therapy. Formations detected during ultrasound decrease when treatment is optimized. Observation of the patient in the dynamics showed that ones of the main diagnostic hormonal tests are blood levels of ACTH and 17-hydroxyprogesterone, which at the time of disease detection were excessively high. Continuous glucocorticoid replacement therapy maintains the level of these indicators within the reference values. Timely diagnosis of the nature of the pathology, constant corrective hormone therapy ensured the patient’s ability to adapt to life and society in accordance with his status. Clinical manifestations of hypocorticism and/or hyperandrogenism in the parents of our patient were not detected, which indicates autosomal recessive inheritance of congenital adrenal hyperplasia. In the future, it is important to provide genetic counseling to expectant parents, especially with manifestations of hyperandrogenism, to assess the possible development of such pathology in their offspring.
адреногенітальний синдром; парціальний гіпокортицизм; гіпогонадизм; крипторхізм; гіперандрогенія; лікування
congenital adrenal hyperplasia; partial hypocorticism; hypogonadism; cryptorchidism; hyperandrogenism; treatment
Вступ
Опис клінічного випадку
/122.jpg)
Висновки
- Pishak V.P., Riznychuk M.O. Adrenogenital syndrome: molecular mechanisms of development. International Journal of Endocrinology (Ukraine). 2017. 2(13). 111-117. (in Ukrainian.)
- Pankiv V.I. Mineralocorticoid deficiency: epidemiology, pathogenesis, diagnosis, treatment approaches. International Journal of Endocrinology (Ukraine). 2012. 3(49). 102. (in Russian.)
- El-Maouche D., Arlt W., Merke D.P. Congenital adrenal hyperplasia. Lancet. 2017, Nov 11. 390(10108). 2194-2210. doi: 10.1016/S0140-6736(17)31431-9. Epub. 2017, May 30. Erratum in: Lancet. 2017. 390(10108). 2142. PMID: 28576284.
- Speiser P.W., Arlt W., Auchus R.J., Baskin L.S., Conway G.S., Merke D.P., Meyer-Bahlburg H.F.L., Miller W.L., Murad M.H., Oberfield S.E., White P.C. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, Nov 1. 103(11). 4043-4088. doi: 10.1210/jc.2018-01865. Erratum in: J. Clin. Endocrinol. Metab. 2019. 104(1). 39-40. PMID: 30272171; PMCID: PMC6456929.
- Nordenström A., Ahmed S., Jones J., Coleman M., Price D.A., Clayton P.E., Hall C.M. Female preponderance in congenital adrenal hyperplasia due to CYP21 deficiency in England: implications for neonatal screening. Horm. Res. 2005. 63(1). 22-28. doi: 10.1159/000082896.
- Strnadová K.A., Votava F., Lebl J., Mühl A., Item C., Bodamer O.A., Torresani T. et al. Prevalence of congenital adrenal hyperplasia among sudden infant death in the Czech Republic and Austria. Eur. J. Pediatr. 2007. 166(1). 1-4. doi: 10.1007/s00431-006-0154-8.
- Kareva M.A., Chugunov I.S. Federal clinical practice guidelines on the management of the patients presenting with congenital adrenal hyperplasia. Problems of Endocrinology. 2014. 60(2). 42-50. (In Russian.) https://doi.org/10.14341/probl201460242-50.
- Melnichenko G.A., Troshina E.A., Molashenko N.V. et al. Russian Association of Endocrinologists clinical practice guidelines for diagnosis, treatment and preventive measures in congenital adrenal hyperplasia due to 21-hydroxylase deficiency patients in adulthood. Consilium Medicum. 2016. 18(4). 8-19.
- Liashuk P.M., Liashuk R.P. Hyperandrogenism syndrome. Chernivtsi: Medical University, 2019. 120 p.